
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
红外光谱、 XPS分析比较Ca2+, Fe3+对石英的活化机理
[刘荣祥 , 李解
, 李解* , 苏文柔, 张雪峰, 李佳伟, 孟留洋]
, 李解, 苏文柔, 张雪峰, 李佳伟, 孟留洋]
|
|


作者简介: 刘荣祥, 1991年生, 内蒙古科技大学硕士研究生 e-mail: 2842014603@qq.com
铁精矿浮选脱硅过程中, 矿浆中的难免阳离子(Ca2+, Fe3+)对阴离子捕收石英的可浮性有重要影响, 而搞清难免阳离子对含石英等脉石矿物的活化机制, 对解决超纯铁精矿脱硅技术难题有重要意义。 目前关于捕收剂对石英吸附结构的研究较多, 而难免离子活化石英的吸附结构及吸附强弱发生机制研究较少。 因此, 采用红外光谱、 XPS检测手段对难免离子(Ca2+, Fe3+)活化石英浮选进行光谱学表征, 同时解析石英中含氧官能团及难免离子的赋存形式, 分析其活化机理。 红外检测结果表明, 在适宜的pH值条件下, Ca2+和Fe3+的加入, 对SDS捕收剂浮选石英均有活化作用, 而活化后的石英与SDS作用, 其间既包括物理吸附, 也包括化学吸附; 而且Fe3+活化作用下的Si—O特征峰红移波数大于Ca2+活化作用下的红移波数, 是由于Ca2+活化石英是单氧-硅键作用, 其键能小, 吸附弱, 而Fe3+活化石英是双氧-硅键作用, 其键能大, 吸附强。 XPS测试表明, Fe3+活化石英的结合能(Fe(2 p)结合能为711.16 eV)强于Ca2+活化石英的结合能(Ca(2 p)结合能为346.93 eV), 其Si(2 s)和Si(2 p)结合能化学位移量更大, 说明Fe3+活化作用下其化学吸附更稳定、 更致密, 且产生两个活性位点, 在石英表面生成稳定的Fe基六元环螯合物; 而对比Fe3+和Ca2+活化作用下的化学吸附不稳定、 不致密, 在石英表面生成Ca基链状络合物。 综合红外光谱、 XPS分析表明, Fe3+比Ca2+有更强的活化作用, 同时加强了药剂与石英表面的化学吸附和物理吸附, 更利于活化石英的浮选。
During the flotation desilication of iron concentrate, the unavoidable cations (Ca2+, Fe3+) in the pulp have important influences on the floatability of quartz using the anion collecter, and it is of great significance to find out the activation mechanism of the unavoidable cations on the quartz and other pulsar minerals to solve the technical problem of desilication of ultra-pure iron concentrate. At present, there are many pieces of research on the adsorption structure of quartz for collectors, while there are few pieces of research on the adsorption structure and the occurrence mechanism of adsorption strength for inevitable ion-activated quartz. So infrared spectroscopy and XPS analysis were adopted, the spectral characterization of unavoidable ions (Ca2+, Fe3+) activated quartz were performed, and the occurrence forms of oxygen-containing functional groups and unavoidable ions in quartz were analyzed, the mechanism of unavoidable ions activated quartz were also analyzed. The results show that in the infrared characterization, at the appropriate pH value, the addition of Ca2+ and Fe3+ activates the flotation of quartz. When quartz is activated by Ca2+ and Fe3+ and reacts with SDS, the chemical adsorption and physical absorption occur almost at the same time. And the red-shifted wave numbers of Si-O characteristic peak under Fe3+ activation is stronger than that of Ca2+ activation. Ca2+ can activate quartz is due to mono- silicon bond, and the bond action has small bond energy and weak adsorption; Fe3+ activated quartz is due to a dioxy-silicon bond, which has large bond energy and strong adsorption. The XPS test results show that the binding energy of Fe3+ activator activated quartz (Fe(2 p) binding energy of 711.16 eV) is stronger than that of Ca2+ activated quartz (Ca(2 p) binding energy of 346.93 eV), which makes the chemical displacement of Si(2 s) and Si(2 p) binding energy larger. It is indicated that the stable Fe-based six-membered ring chelate is formed on the surface of quartz under the activation of Fe3+, and chemical adsorption is more stable and dense, and two active sites are generated; while the unstable Ca-based s chain-like complex is formed on the surface of quartz under the activation of Ca2+, and chemical adsorption is unstable and not so dense. Comprehensive infrared spectrum and XPS analysis show that Fe3+ has stronger activation than Ca2+, and enhance the chemical and physical adsorption between the agent and quartz surface, which is more conducive to the flotation of activation quartz.
我国的铁矿普遍存在“贫、 细、 杂”的特点, 在生产超纯铁精矿过程中, 为了降低石英(SiO2)的含量, 通常采用“磨矿、 磁选、 浮选”等联合工艺达到预期指标[1]。 其中, 反浮选脱硅工艺是提铁降硅的关键步骤[2], 因此, 研究药剂(尤其是活化剂)与石英的相互作用机理至关重要, 而红外光谱、 XPS分析是研究药剂与矿物之间作用的常用手段。 有报道曾用红外光谱分析来判定药剂在矿物表面作用的三种机制, 判定标准为: 药剂作用后的矿物谱图中如有新的吸收峰, 为化学反应; 如发现吸收峰的位置红移, 且超出设备误差范围, 属于化学吸附; 而通过反复水洗矿物表面沾染的药剂后, 矿物红外谱图中未发现药剂分子, 则药剂在矿物表面发生的是物理吸附。 而目前关于金属阳离子浮选活化石英的机理至今没有统一定论, 主要用金属阳离子在石英表面形成了羟基络合物[3]或金属氢氧化物沉淀[4]活性位点来解释活化后的石英与捕收剂发生吸附加强了石英的可浮性。 从金瑶等[5]研究发现, Ca(OH)+是油酸钠体系下Ca2+活化石英的主要活性成分, 而Ca(OH)2沉淀是不利于石英浮选的主要成分, 陈琳璋等[6]也表明在阴离子捕收剂SDS体系中Ca2+在适宜的pH值下对石英有活化作用, Ca(OH)+同样是Ca2+活化石英的主要活性成分, 发生物理吸附而有利于阴离子捕收石英; 但郭玉[7]却认为在石英表面的Ca(OH)2与阴离子捕收剂发生反应, 加强了物理吸附, 从而有利于石英浮选。 而对于铁离子的活化, 欧乐明等[8]发现油酸钠体系下的Fe3+和Fe2+在碱性条件下能活化石英的浮选; Zhou等[9]的红外光谱分析表明Fe3+对石英的活化, 有利于捕收剂SDS捕获石英。 除了Ca2+和Fe3+, 其他阳离子也对石英浮选有很好的活化作用[10, 11], 高跃升等[12]总结了各种金属离子对矿物浮选行为影响规律和作用机理表明金属离子与OH-的结合, 包括金属离子羟基配合物和金属氢氧化物表面沉淀, 它们可能同时存在。 另外, 史学伟[13]采用XPS分析mSiO2-IDA对Cu2+和Cd2+的吸附过程, 发现其吸附机制并不相同。 Ruan研究了Ca2+对磷灰石、 白云石和石英浮选的影响, 通过XPS分析发现Ca2+能增强阴离子捕收剂对磷灰石、 石英的吸附, 而对浮选白云石的影响并不明显[14]。
基于前人研究, 本试验选用Ca2+和Fe3+为活化剂, 通过对石英与活化剂、 捕收剂作用前后的红外光谱、 XPS检测分析, 研究Ca2+和Fe3+对石英的活化机理。
纯矿物石英来源于包头白云鄂博, 经手选, 破碎, 锆石球磨机细磨, 获得粒径为-0.074~0.038 mm, 纯度为99.5%的石英粉末。 化学试剂有FeCl3, CaCl2, HCl, NaOH和十二烷基磺酸钠(SDS), 均为分析纯; 实验用水为去离子超纯水。 采用PHS-3型酸碱度pH计(上海科学仪器公司)测量pH值。
具体制备流程: 将一定量的石英粉末分别倒入1#, 2#和3#烧杯中, 再各加50 mL去离子水, 其中1#加入捕收剂SDS(90 mg), 调节pH值为9; 2#加入活化剂CaCl2(90 mg)+SDS(90 mg), 调节pH值为12; 3#加入活化剂FeCl3(90 mg)+SDS(90 mg)的药剂, 调节pH值为7, 在室温下磁力搅拌机上搅拌30 min, 离心过滤, 取出滤渣, 水洗、 室温下干燥12 h, 最终得到待测样品。
采用VT-70型傅里叶变换红外光谱仪(BURKER公司)检测石英样品作用前、 后的红外光谱, 波数为4 000~400 cm-1, 分辨率为1 cm-1, 扫描速度为2.5 kHz, 采用光谱纯的KBr作为载体, 样品与KBr按1:200混合研磨至5 μm, KBr压片法测定; 采用美国Thermo Fisher Scientific公司生产的ESCALAB2250Xi型光电能谱仪(XPS)对石英样品作用前、 后进行XPS测试, 检测条件: Al Kα 激发源, 靶电压和靶电流分别为15 kV和10 mA, 真空室气压< 2×10-6 Pa, 分析器传输能量为50 eV, 测量步长为0.1 eV, 溅射速度为0.2 nm·s-1, 溅射面积为2 mm×2 mm; 采用Avantage v5.52专用软件在计算机上执行对校正后的能谱数据进行泰勒分峰拟合, 所需XPS标准谱线数据引自XPS手册。
先采用Ca2+和Fe3+活化石英, 再加SDS捕收剂, 对石英与药剂作用前后的样品进行红外光谱分析, 结果如图1所示。
 | 图1 石英与药剂作用前、后的红外光谱 (1): SDS; (2): 石英; (3): 石英+SDS; (4): 石英+Ca2++SDS; (5): 石英+Fe3++SDSFig.1 FTIR spectra of quartz and reagents before/after interaction (1): SDS; (2): Quartz; (3): Quartz treated with SDS; (4): Ca2+ activated quartz treated with SDS; (5): Fe3+ activated quartz treated with SDS |
红外光谱[图1曲线(1)]是捕收剂SDS的红外光谱, 其在3 438 cm-1处的吸收峰是—OH吸收峰, 并且在2 928和2 857 cm-1处是—CH2—, —CH3吸收峰[15], 1 446 cm-1处是—CH2—吸收峰, 吸收带磺酸盐的特征峰出现在1 200~950 cm-1[15], 其中1 190和1 042 cm-1处的吸收峰是磺酸盐的—S
当pH值为12时, 石英经Ca2+活化后与SDS作用, 其红外光谱显示如图1的曲线(4)所示; 而当pH值为6, 石英经Fe3+活化后与SDS作用, 其红外光谱如图1中曲线(5)所示。 从中可以看出, —CH2—和—CH3的伸缩振动吸收峰出现在曲线(4)和(5)中的2 931和2 857 cm-1处, 对比SDS捕收剂[图1中曲线(1)], 其—CH2—和—CH3伸缩振动吸收峰的峰位并无波数的移动, 符合物理吸附的特性, 但特征峰强度增强, 而且Fe3+活化作用下吸收峰的强度比Ca2+的强, 说明Fe3+活化作用下发生的物理吸附聚集的SDS药剂官能团更多, 结构更致密; 而Si—O振动吸收峰分别出现在曲线(4)和(5)中的1 087和1 095 cm-1处, 与曲线(2)在1 085 cm-1处的Si—O相比, 波数分别正红移2和10 cm-1, 同时峰的强度和面积增大, 说明石英与活化剂之间发生化学吸附, 且Fe3+的化学吸附作用强于Ca2+, 表面吸附有机药剂的基团数目更多[15]。
由于红外光谱仪的分辨率为1 cm-1, 故Ca2+和Fe3+活化后出现波数正红移不是仪器误差, 而且由公式[15]ν =
综上分析, 可知Ca2+和Fe3+与石英的作用为化学吸附, SDS与Ca2+和Fe3+之间为物理吸附; Ca2+和Fe3+的活化既强化了物理吸附, 也强化了化学吸附, 从而利于SDS捕收石英, 且Fe3+的活化作用强于Ca2+。
为了进一步确定石英与药剂作用的表面吸附结构, 对石英与药剂作用前后的样品进行XPS分析。 结果见表1、 图2。
| 表1 石英与不同药剂作用前后元素的原子轨道结合能和原子数分数 Table 1 Atomic orbital binding energy and atomic fraction of elements before and after the action of quartz and different agents |
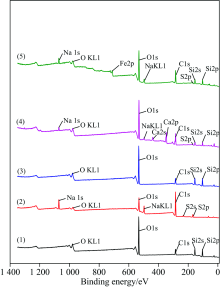
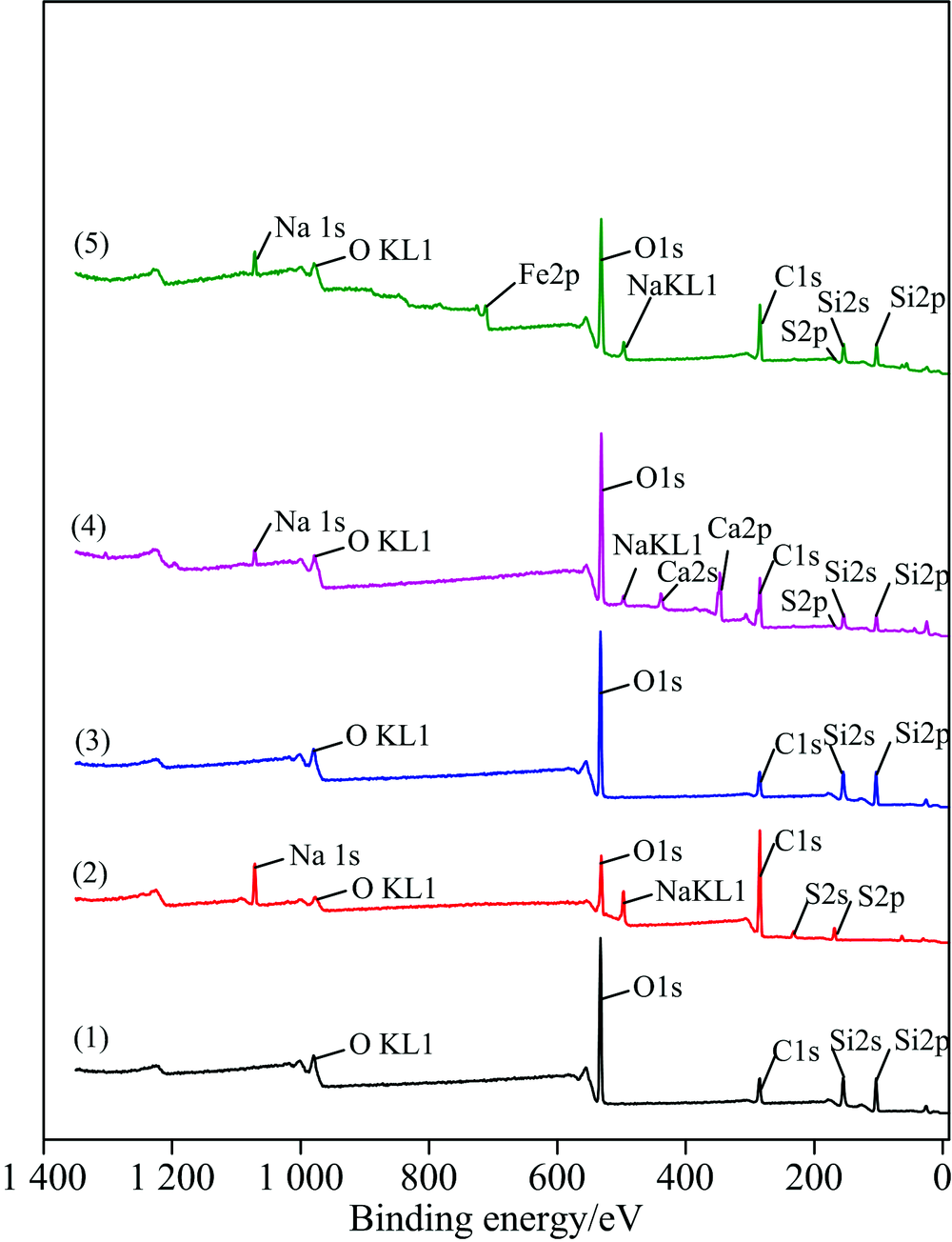 | 图2 经药剂处理前后石英的XPS全谱图 (1): 石英; (2): SDS; (3): 石英+SDS; (4): 石英+Ca2++SDS; (5): 石英+Fe3++SDSFig.2 XPS full spectrum of quartz before and after drug treatment (1): Quartz; (2): SDS; (3): Quartz treated with SDS; (4): Ca2+ activated quartz treated with SDS; (5): Fe3+ activated quartz treated with SDS |
图2(1), (2), (3), (4)和(5)所示分别为石英、 SDS、 石英与SDS作用后、 石英经Ca2+活化与SDS作用、 石英经Fe3+活化与SDS作用后的XPS能谱。
石英全谱如图2曲线(1), 其中有Si(2s), Si(2p), O(1s), OKL1, C(1s), 表明石英为纯矿物没有金属离子的干扰; SDS全谱如图2曲线(2), 其中有S(2s), S(2p), Na(1s), NaKL1, O(1s), OKL1, C(1s); 石英与SDS作用的XPS峰如图2曲线(3), 与图2曲线(1)中石英的XPS峰相比, 发现未出现新的峰, 但是各元素的结合能和元素百分数有所变化。 由表1可知, 石英与SDS作用之后与作用之前相比, Si(2p)结合能改变为0.03 eV, Si(2s)结合能改变为0.2 eV, O(1s)结合能改变为0.01 eV, C(1s)结合能改变为0.04 eV, 位移值均在仪器误差(0.3 eV)范围之内, 说明单纯捕收剂SDS对石英内层电子的结合能影响很低, 由此推断单纯使用捕收剂SDS, 不会对石英发生吸附作用。
石英经Ca2+活化与SDS作用如图2曲线(4), 从谱线中可以看出, 有新的峰出现, 即Ca(2p), S(2p), Na(1s)和NaKL1, 说明发生吸附作用。 由表1可知, Ca2+活化石英后, 石英表面Ca(2p)结合能为346.93 eV, 其元素分数为6.55%; 而Si(2s)结合能改变值为0.74 eV, 元素分数为9.3%, Si(2p)结合能改变值为0.62 eV, 元素分数为9.85%; O(1s)结合能改变值为1.26 eV, 元素分数为33.64%, 相比原矿O(1s)元素分数下降了1.34%, C(1s)结合能改变值为0.44 eV, 元素分数为30.67%, 相比原矿C(1s)元素分数上升了13.67%; Si(2s)和Si(2p)的元素分数相比石英中的Si(2s)和Si(2p)大幅度下降, 这是含Si官能团可能与Ca2+发生了配位反应, Si(2s)和Si(2p)原子轨道的几何形状有可能发生改变, 说明石英表面存在Ca2+的化学吸附, 并形成Si—O—Ca(OH)键, 而Si—O—Ca(OH)键与SDS中的S(2p)发生物理吸附, 且SDS的外层为疏水性, 故易于浮选。
石英经Fe3+活化与SDS作用如图2曲线(5), 从谱线中可以看出有新峰出现, 即Fe(2p), S(2p), Na(1s)和NaKL1, 表明发生了吸附。 由表1可知石英表面Fe(2p)结合能为711.16 eV, 其元素分数为2.05%。 经Fe3+活化石英的XPS图谱中Si(2s)结合能改变为0.86 eV, 元素分数为12.55%, Si(2p)结合能改变为0.79eV, 元素分数为14.17%, 这与Fe3+发生的配位反应使Si(2s)和Si(2p)原子轨道的几何形状发生改变有关, 而O(1s)结合能改变为1.06 eV, 元素分数为30.75%, 相比原矿O(1s)元素分数下降了4.23%, 这与O原子周围电子云密度的增加使石英与Fe3+发生吸附效应有关, C(1s)结合能改变为0.59 eV, 元素分数为30.51%, 相比原矿C(1s)元素分数上升了13.51%, Si(2s)和Si(2p)的元素分数相比石英中的Si(2s)和Si(2p)大幅度下降, 说明石英表面存在Fe3+的化学吸附, 并形成键, 且键与SDS中的S(2p)发生物理吸附。 因为在如图2曲线(5)中S(2p)元素分数为1.38%, 比图2(4)活化石英时S(2p)元素分数高, 而SDS的外层为疏水性, 易浮选, 相比较图2(4)而言, Fe3+活化石英的结合能[Fe(2p)结合能为711.16 eV]强于Ca2+的结合能[Ca(2p)结合能为346.93 eV], 经Fe3+活化石英的XPS图谱中Si(2s)和Si(2p)结合能化学位移量更大, 表明Fe3+活化石英比Ca2+活化石英更稳定, 使SDS在石英表面生成稳定的螯合物, 增强了捕收剂对石英表面的吸附作用。
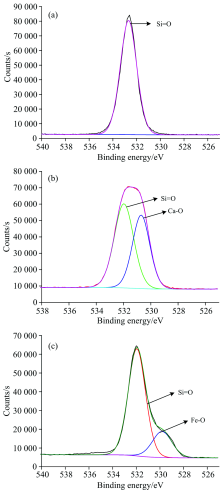
图3为活化剂活化前后的石英与SDS作用的O(1s)XPS高分辨谱。 由图3(a)可知, O(1s)峰主要来源为石英, 峰型单一且比较对称, 光电子强度和峰面积最大, 存在Si=O的O(1s)峰, 位于532.68 eV, 光电子强度为77 935.01 Counts·s-1, 峰面积为137 958.82 CPS·eV。 说明石英很纯, 没有其他杂质的影响。 与Ca2+活化剂作用后的图3(b)相比, O(1s)峰对称性降低, 经化学状态分析和泰勒分峰, O(1s)峰存在Si=O—Ca—O的O(1s)峰, Si=O的O(1s)峰位于531.97 eV, 发生谱峰位移, 光电子强度降低至51 352.65 Counts·s-1, 峰面积降低至93 159.35 CPS·eV, 这是因为羟基络合物上的氧元素稀释了Si=O的O(1s)峰的相对含量; 而出现的Ca—O的O(1s)峰位于530.71 eV, 光电子强度为44 893.74 Counts·s-1, Ca—O的O(1s)峰面积为73 132.91 CPS·eV; 可以看出活化的石英表面O原子与金属离子Ca2+发生化学吸附, 致使O(1s)峰型发生变化, 失去了对称性, 并生成了含Si—O—Ca键的物质。 而Fe3+活化剂作用后见图3(c), O(1s)峰对称性降得更低, 存在Si=O/Fe—O的O(1s)峰, Si=O的O(1s)峰位于531.95 eV, X射线能谱线位移为0.73 eV, 较Ca2+活化剂作用后Si=O的O(1s)峰位移量大, 光电子强度降低至57 217.54 Counts·s-1, 且峰面积降至107 121.86 CPS·eV; 又出现了Fe—O的O(1s)峰位于529.82 eV, 光电子强度为13 877.89 Counts·s-1, 峰面积为29 272.38 CPS·eV; 可以看出活化表面O原子与金属离子Fe3+发生化学吸附, 导致O(1s)峰型变化, 失去了对称性, 同样生成了Si—O—Fe键的物质。
 | 图3 活化前后的石英与SDS作用的O(1s) XPS高分辨谱 (a): 石英; (b): 石英+Ca2++SDS; (c): 石英+Fe3++SDSFig.3 O(1s) XPS high resolution spectrum of quartz and SDS before and after activation (a): Quartz (b): Ca2+ activated quartz treated with SDS; (c): Fe3+ activated quartz treated with SDS |
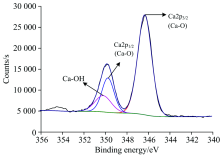
图4为石英与活化剂作用后的石英Ca(2p) XPS高分辨谱。 经过活化剂作用, 分别出现了Ca(2p)的新峰, 经过数据处理和泰勒分峰, 确定了石英表面膜中金属原子的氧化状态, Ca(2p)存在Ca(2p)3/2(位于346.73 eV, 光电子强度为23 751.99 Counts·s-1, 峰面积为39 507.47 CPS·eV)、 Ca(2p)1/2(位于350.13 eV, 光电子强度为9 347.86 Counts·s-1, 峰面积为13 503.11 CPS·eV)的Ca—O键的峰和Ca—OH键(位于350.80 eV, 光电子强度为3 343.69 Counts·s-1, 峰面积为5 778.41 CPS·eV)的峰, 从Ca(2p)存在的峰面积可以看出Ca(2p)3/2/ Ca(2p)1/2/Ca—OH含量比约为4:3:1, 说明在石英表面有Ca的羟基络合物生成, 氧原子与一价羟基络合物脱水生成Si—O—Ca—OH。
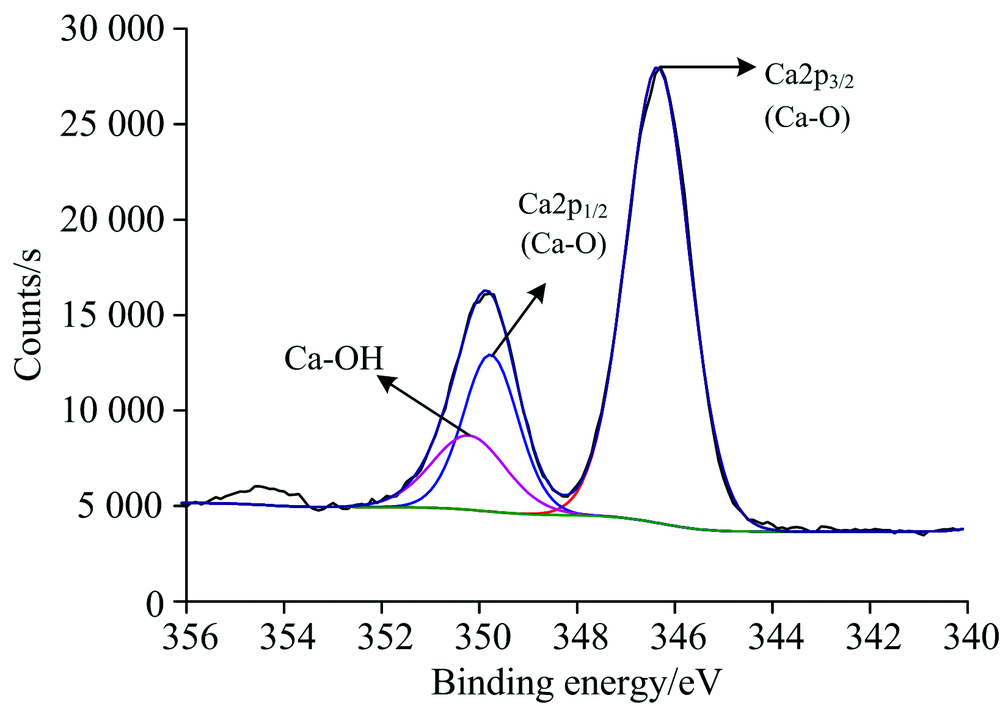 | 图4 活化剂作用后的石英Ca(2p)高分辨谱Fig.4 High resolution spectrum of quartz Ca(2p) after activator |
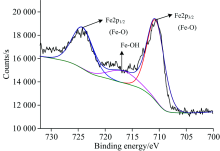
图5为石英与活化剂作用后的石英Fe(2p)XPS高分辨谱。 由图可见, 经过活化剂作用, 分别出现了Fe(2p)的新峰, 经过数据处理和泰勒分峰, Fe(2p)存在Fe(2p)3/2(位于710.79 eV, 光电子强度为7 266.86 Counts·s-1, 峰面积为26 474.75 CPS·eV)、 Fe(2p)1/2(位于724.23 eV, 光电子强度为3 662.09 Counts·s-1, 峰面积为13 354.29 CPS·eV)的Ca—O键的峰和Fe—OH键(位于718.00 eV, 光电子强度为1 740.43 Counts·s-1, 峰面积为6 346.32 CPS·eV)的峰, 从Fe(2p)存在的峰面积可以看出Fe(2p)3/2/Fe(2p)1/2/Fe—OH含量比约为3:2:1, 说明在石英表面有铁的羟基络合物生成, 氧原子与一价羟基络合物脱水生成Si—O—Fe—OH。
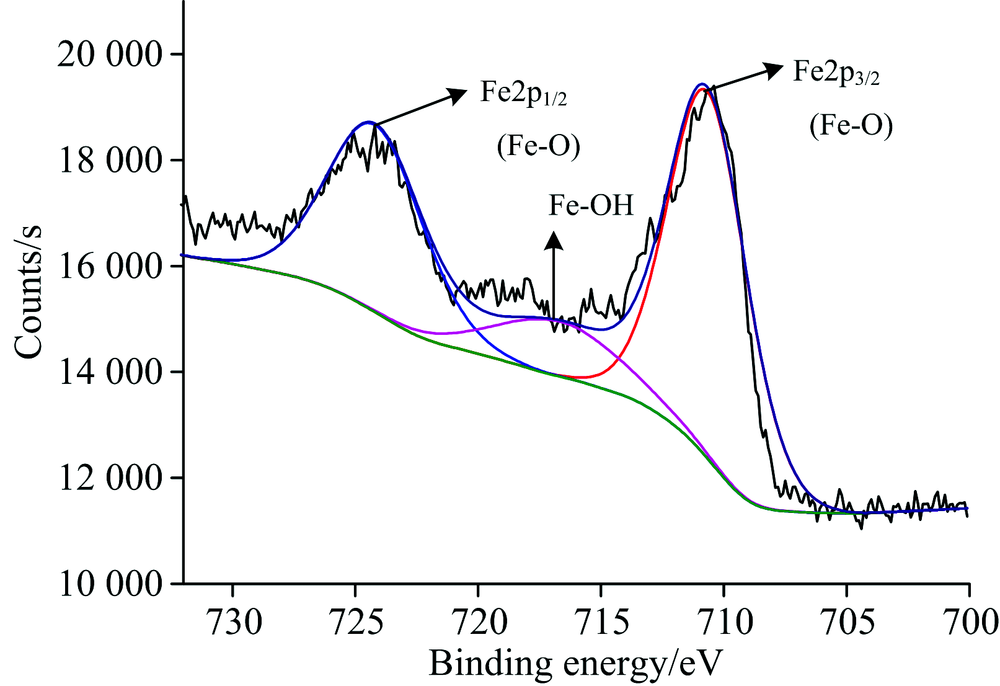 | 图5 活化剂作用后的石英Ca(2p)高分辨谱Fig.5 High resolution spectrum of quartz Ca(2p) after activator |
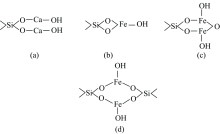
当Ca2+活化石英时, pH值等于12时, 石英的羟基失去质子, 以—O-与一价羟基钙配位, 生成Si—O—Ca—OH键, 成为石英的活化位点, 再与阴离子进行物理吸附, 可能的构型如图6(a), 这样的结构式为链状结构, 不太稳定。 以三价铁在pH值等于7时, 优势组分为Fe(OH)3沉淀, 而石英上的一个羟基失去质子, 沉淀物的羟基发生脱水反应, 生成Si—O—Fe—OH键, 与阴离子捕收剂进行物理吸附, 可能的构型如图6(b), (c)和(d), 但是, 若以三价的铁与石英表面的—O-配合, 则生成四元螯合物合物, warner指出四元环螯合物比较少, Ley指出五元环或者六元环的螯合物最为稳定, 图6(c)和(d)所示的两种螯合物合物, 使的石英矿物表面出现金属-氧化物的O(1s)峰。
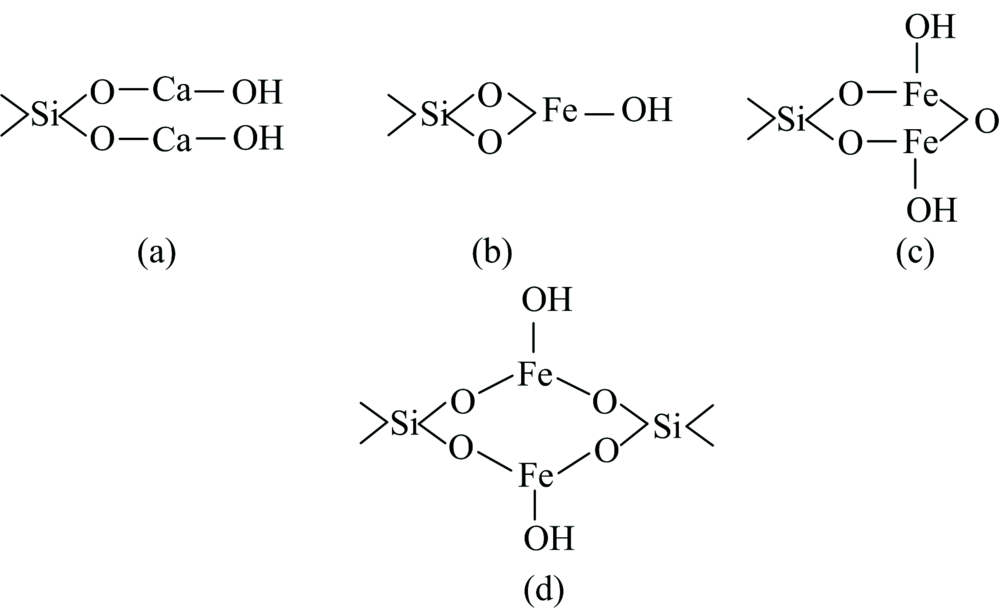 | 图6 活化剂与石英发生吸附可能存在的(a, b, c, d)四种构型Fig.6 Possible four configurations (a, b, c, d) configuration of activator and quartz adsorption |
在参考其他研究者的矿物与药剂作用时, 常常提及“因为四环张力太大, 从而不稳定”, Adols Von Bayer说基于两点[16]: (1)所有环状化合物都具有平面结构, (2)键角与sp3杂化轨道正常键角(109° 28')有差别。 实际上多元环是比较稳定的, 原因再与Bayer学说第一点假设, 目前已通过核磁共振等手段证明, 多元化合物非严格的平面结构, 所以多元螯合物更稳定。 对比图6活化剂与石英发生吸附可能存在的构型, (a)为Ca—O—Si的链状结构, 最不稳定; (b), (c)和(d)为Fe—O—Si环状结构, 其F(2p)3/2结合能为710.29 eV, Fe(2p)1/2结合能为724.23 eV, 所形成的螯合物较为稳定, 但(b)为四元环, 张力太大, 不稳定; 而若环小于4而大于7时, 形成闭合环的可能性下降, 故(d)结构不存在; 而对于共轭的配位体, 六元环(C)结构最稳定[17]。
因此, 当使用Ca2+活化剂时, 最有可能的是(a)构型; 当使用Fe3+活化剂时, 最有可能的是(c)构型。
(1)FTIR测试表明, 纯矿物石英经阳离子(Ca2+, Fe3+)活化并与SDS捕收剂作用后, 石英的红外光谱中, 新增—CH2—和—CH3的伸缩振动的吸收峰, 但没有波数移位, 在石英表面发生物理吸附; 而Si—O非对称伸缩振动吸收峰有波数的正向偏移和吸收峰的增强, 键能增大, 又表明在石英表面发生了化学吸附。 而且, Fe3+活化作用下的特征吸收峰强度明显强于Ca2+活化时的强度, 是由于Ca2+活化时, 吸附为一价羟基络合物的单氧键合作用, 其键能小, 吸附作用弱; Fe3+活化时, 吸附为Fe(OH)3沉淀的螯合作用, 为双氧键合作用, 它的键能是大的, 吸附作用强, 因此在适宜的pH值下, Fe3+比Ca2+表现出更强的活化作用, 加强了SDS在石英表面的吸附, 更利于活化石英的浮选。 Ca2+和Fe3+作用在石英表面属于化学吸附, 而SDS吸附在活化剂作用之后的石英上为物理吸附。
(2)XPS测试表明, Fe3+活化剂活化石英的结合能(Fe(2p)结合能为711.16 eV)强于Ca2+活化石英的结合能(Ca(2p)结合能为346.93 eV), 其在XPS图谱中使Si(2s)和Si(2p)结合能化学位移量更大, 说明活化作用下的Fe3+与石英的化学吸附更稳定、 更致密, 且在石英表面既生成稳定的Fe基六元环螯合物, 产生两个活性位点, 从而证明Ca2+和Fe3+活化剂与石英之间的化学吸附加强, 而活化之后的石英与SDS间的物理吸附。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|