

{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于参数校正的近红外光谱模型转移新方法
[胡芸1  , 李博岩
, 李博岩2, * , 张进2 , 彭黔荣1 ]
, 李博岩, 张进|
|


作者简介: 胡 芸, 1978年生, 贵州中烟工业有限责任公司技术中心工程师 e-mail: huyunyun99@hotmail.com
模型转移是解决近红外光谱仪器间存在差异导致校正模型难以在多台仪器间通用问题的重要方法。 利用主成分-马氏距离方法判断样品在不同仪器间的光谱差异性, 然后通过吉洪诺夫正则化约束和校正模型参数, 提出新的模型转移算法, 实现模型在不同近红外光谱仪器上的共享和使用。 首先使用一组标准样品光谱, 建立主机和子机近红外光谱模型预测误差最小化函数。 通过约束主机和子机的模型参数的差异, 求出子机的模型参数, 从而达到模型转移的目的。 该方法应用于药物活性成分和烟叶中总植物碱与总糖的含量分析, 结果表明使用15个标准样品时, 子机光谱样本的预测均方根误差(RMSEP)分别从8.3 mg、 0.49%和1.91%降到3.9 mg、 0.09%和0.83%。 转移后模型预测相对分析误差(RPD)均大于3.0, 子机光谱样本的预测效果得到明显提高。 该方法理论明确、 直观, 在实际应用中样品预测准确性较好, 为具有标准样品的模型转移方法提供一种新思路。
Model transfer plays an important role in solving the problem of the difference between near infrared (NIR) spectroscopic instruments and the prediction difficulty of models. The spectral differences between the same samples taken on different NIR instruments were identified using the principal component-Mahalanobis distance method. Based on the constrain conditions of Tikhonov regularization (TR) and model parameter correction, a new algorithm (called new Tikhonov regularization-based calibration transfer, NTRCT) was proposed for calibration transfer between NIR instruments, so as to facilitate the share and use of the calibration models. The spectra of a set of standard samples were first utilized to establish a specific function that could minimize the prediction errors obtained from the master and slave instrumental models. By constraining the difference of the model parameters, the parameters of the slave instrument model were then determined, to achieve the purpose of model transfer from the master instrument to the slave one. This method was applied to analyze the content of the active pharmaceutical ingredient (API) of tablets and quantify the contents of total alkaloids and total sugars in tobacco leaves respectively, by means of their NIR spectra acquired on different instruments. The results showed that the root means square error of prediction (RMSEP) of samples taken on the salve instrument was reduced from 8.3 mg, 0.49% and 1.91% to 3.9 mg, 0.09% and 0.83% respectively; when 15 standard samples were employed for modelling. As the calibration transferred all the resulting RPD values were larger than 3.0, and the sample predictions from the salve instrumental spectra were thus significantly improved. The method was explicit and intuitive in theory, and had good accuracy in sample prediction in practical applications. It provided a new idea for calibration transfer method with standard samples.
近年来, 基于近红外光谱的快速检测分析迅速发展并被广泛应用于烟草、 石油等领域。 近红外光谱分析主要是通过收集大量样品数据建立多元校正模型, 进而实现组分的定性和定量分析目的。 与常规检测方法相比, 具有无损、 绿色、 简单快捷等优点。 在实际应用过程中, 由于近红外仪器的更新、 维修、 老化, 或不确定的外界因素等变化会引起光谱数据的改变, 从而导致校正模型的预测能力降低或者根本不能使用。 因此, 为了提高模型预测的准确性和适用范围, 研究与应用合理的模型转移方法就显得尤为重要。
模型转移的主要思路是建立主机(master instrument)和子机(slave instrument)光谱、 模型参数或预测值之间的函数关系, 进而校正由于仪器或检测环境因素变化导致的样本预测误差[1, 2, 3]。 按照校正的对象不同, 模型转移方法大致可以分为三类: (1)对预测结果进行校正, 如模型斜率/截距(S/B)修正算法等[4]; (2)对光谱进行校正, 如分段直接标准化(piecewise direct standardization, PDS)等[5, 6, 7, 8]; (3)对模型参数进行校正, 如两步偏最小二乘方法等[9]。 模型参数校正方法简单、 实用, 不涉及近红外光谱的校正。 当然, 不同的分析体系所适用的模型转移方法会不同, 转移模型的预测准确性也有差异。
正则化(regularization)是一种通用的防止参数过拟合的方法。 在化学计量学回归和聚类算法中广泛使用不同的正则化约束方法[10], 如岭回归、 LASSO和弹性网等。 本文基于吉洪诺夫正则化提出了一种参数校正的模型转移新方法(new Tikhonov regularization-based calibration transfer method, NTRCT)。 其思路是通过同时约束主机与子机光谱模型, 使得标准样品的主机与子机模型的预测差异最小。 该方法为有标准样本的模型转移方法, 简单、 直接。 将该方法分别应用于药物和烟叶的近红外光谱数据分析, 其偏最小二乘模型转移效果令人满意。
药物的透射近红外光谱数据采自于两台近红外仪器(Foss NIR systems, Silver Spring, MD), 来源于国际漫反射会议网(http://www.idrc-chambersburg.org/shootout2002.html)。 光谱的波长范围为600~1 898 nm, 间隔为2 nm, 药物的有效成分(API)含量范围为151.6~239.1 mg。 样本集包含655个样本, 其中校正集有155个样本, 验证集40个样本, 预测集460个样本。 依据文献[6], 剔除4个校正集异常样本(即#19, 122, 126和127样本), 9个预测集异常样本(#11, 145, 267, 294, 295, 313, 341, 342和343样本)。
烟叶的近红外光谱采自两台Thermo Antaris Ⅱ 傅里叶近红外分析仪器(Thermo Scientific公司)。 光谱的波数范围为10 000~4 000 cm-1, 分辨率为8 cm-1, 扫描次数为64。 按照烟草及烟草制品总植物碱与水溶性糖的测定标准, 采用连续流动分析法测得烟叶中总植物碱含量范围为1.28%~3.96%, 总糖含量范围为7.92%~36.18%。 利用Kennard-Stone算法对209个烟叶样本的光谱进行选样, 40个样本作为标准样品, 120个样本用作校正集, 剩余的49个样本作为测试集。
样品在主机和子机上量测所得近红外光谱分别为Xm和Xs, 目标组分的偏最小二乘定量校正模型可表示
不同仪器采集相同样品的近红外光谱之间存在差异性, 但目标组分的含量是一致的, 因此, 使用主机和子机光谱模型对样品进行预测时, 其差异可用式(3)表示
这里, 我们定义e为模型预测损失函数。
若光谱存在微小线性差异, 模型参数差异则较小, 可以使用相关系数corr(β m, β s)> rth作为约束优化模型[11]。 化学计量学中常使用稀疏或者平方等正则化约束。 其中平方约束能够有效地约束向量之间的夹角和长度, 是一种性质优异的约束条件。 因此, 定义主机和子机光谱模型参数的平方约束小于一个特定值ξ ,
方程(3)中平方损失函数在式(4)的约束下, 可以转化为最小化损失函数
其中λ为权重参数, 决定着子机光谱模型的预测准确度和模型复杂度。 当λ较小时, 使用不同仪器模型预测样品的差异最小化占主导作用, 而忽略了对模型一致性的约束, 结果可能导致模型过拟合(over-fitting); 当λ过大时, 则过分强调模型一致性, 从而导致子机预测结果变差, 引起欠拟合(under-fitting)问题。 通过对式(5)中的损失函数求导数, 并令其等于零, 得到该损失函数的极小值
其中I表示单位矩阵。 该方法简单、 稳健, 能够直接求解出最优解, 无需优化算法。
采用偏最小二乘(partial least squares, PLS)方法建立校正模型; 利用交互检验均方根误差(root mean square error of cross validation, RMSECV)、 预测均方根误差(root mean square error of prediction, RMSEP)和相对分析误差(relative prediction deviation, RPD)(即建模数据分布标准偏差与预测均方根误差的比值)[12]三个指标评价模型建立和转移效果。 RPD综合考虑预测样本化学值的标准差与所建模型的预测标准差, 是评价模型分辨能力的重要参数。 通常, RPD> 3.0, 说明定标效果良好, 所建模型可用于实际样品检测; RPD=2.5~3.0, 说明所建模型可进行定量分析, 但精度有待提高; RPD< 2.5, 说明该成分定量分析困难。
同一药物样本分别在两台仪器上量测的光谱相似, 而在600~750和1 650~1 800 nm区间内差异较为明显[图1(a)]。 由于药物的近红外光谱在1 750~1 898 nm区间内存在严重的噪声干扰, 因此, 仅选择600~1 738 nm波长范围的光谱信号用于建立模型。 而相同的烟叶样本在两台仪器上测量所得的光谱有一定的背景差异, 但整体形状非常相似[图1(b)]。
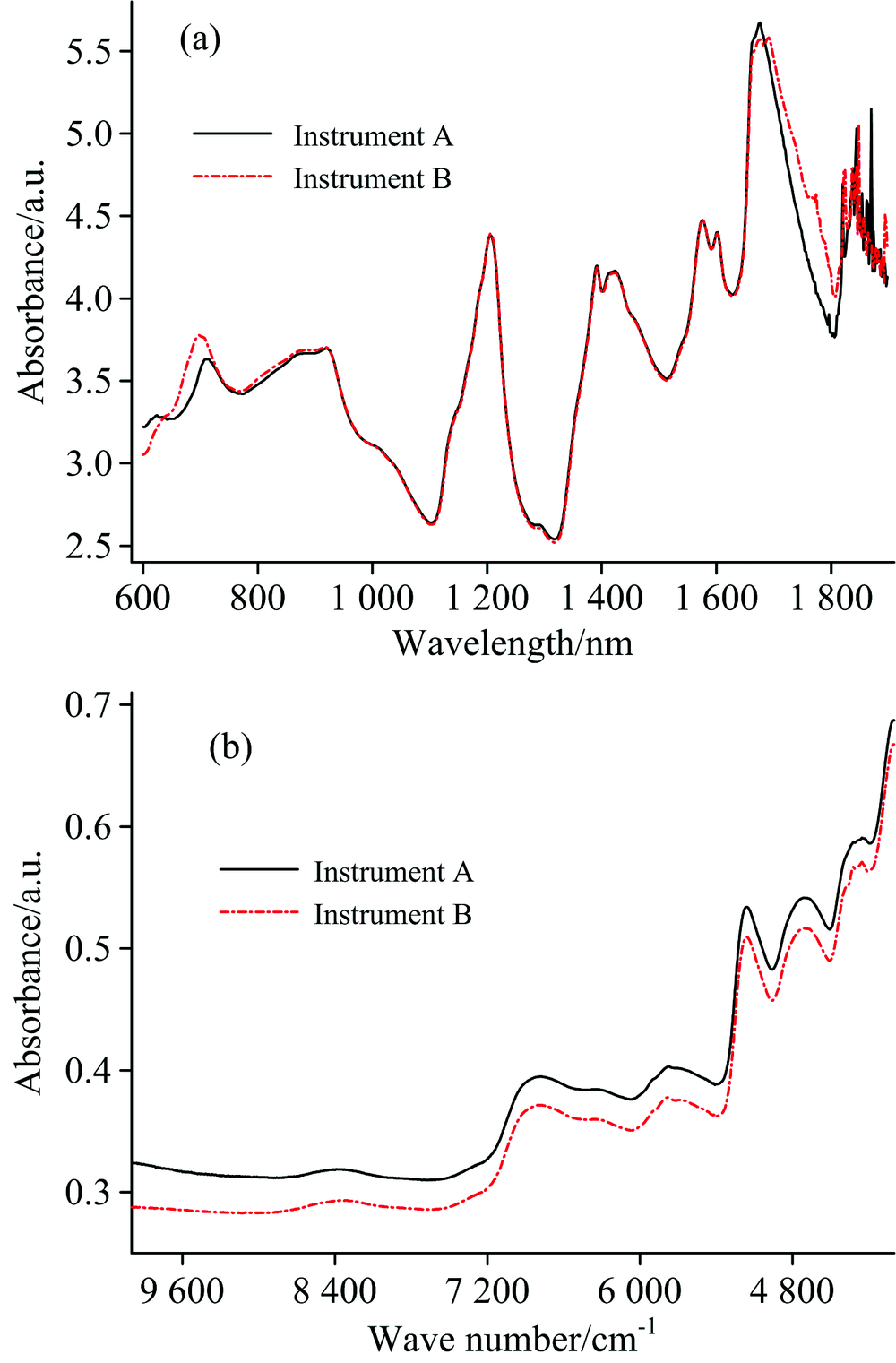 | 图1 同一药物(a)和烟叶(b)样本在两台仪器上量测的近红外光谱图Fig.1 NIR spectra of the same sample collected on two instruments (a) pharmaceutical tablet and (b) tobacco leaf |
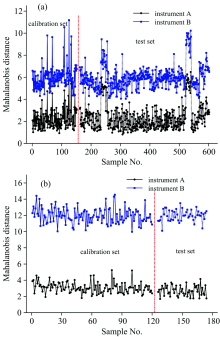
我们采用主成分-马氏距离(PCA-Mahalanobis)方法, 提取样品光谱的特征信息, 进而表征同一样本体系在不同仪器上的光谱性质或特征的相似程度。 无论是就药物还是烟叶样品而言, 在主机上量测样品光谱间的马氏距离小于子机与主机样品光谱间的马氏距离(图2), 例如, 药物和烟叶的测试集样品在主机上量测所得光谱间的马氏距离平均值分别为2.34和2.55, 而在子机上量测光谱与主机光谱间的马氏距离平均值分别为4.69和5.58。 马氏距离的大小一定程度上量化了同一样品在主机和子机上所采集的光谱数据的差异性, 因此直接用主机校正模型预测子机光谱样品参数, 必然会引起预测结果的误差。
 | 图2 (a)药物和(b)烟叶样本在两台仪器上的马氏距离Fig.2 Mahalanobis distance of the samples taken on two instruments: (a) pharmaceutical tablets and (b) tobacco leaves |
NTRCT方法主要是通过对标准样品的预测来优化参数λ, 调整主机和子机光谱预测结果一致性和模型相似性。 λ值的大小对模型转移效果非常关键: λ较小, 则会出现模型过拟合, 过分追求样本的预测效果; λ过大, 则会出现模型欠拟合, 过分强调主机和子机模型的相似程度。 我们以药物样本集为例, 应用15个标准样品, 以子机光谱样本的PLS定量模型的RMSECV为目标, 优化参数λ。 图3(a)给出了药物样本活性成分模型中λ随子机光谱样本的RMSECV变化曲线, 结果表明当λ=2.442时, 其对应的RMSECV为最小, 子机光谱样本预测集的RMSEP为3.9 mg, 接近主机模型预测主机光谱样本的RMSEP 3.4 mg, 大大低于主机模型直接预测子机光谱样本的RMSEP 8.3 mg。 图3(b)和(c)为烟叶中总植物碱和总糖含量模型中参数λ随子机光谱样本的RMSECV变化曲线。 当λ分别为222.300和0.027时, 其对应的RMSECV为最小, 相应的子机光谱预测集样本的总植物碱和总糖的RMSEP为0.09%和0.83%。 该结果表明, 通过选择合适的参数λ, 优化子机模型的参数, 能够提高子机光谱样本的预测结果。
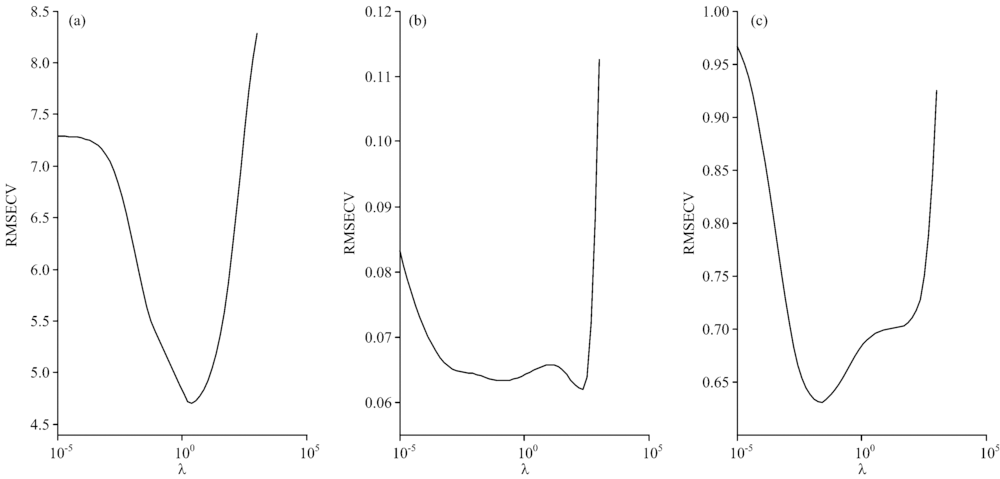 | 图3 (a)药物活性成分、 烟叶样本中(b)总植物碱和(c)总糖的RMSECV随参数λ的变化情况Fig.3 Effect of the parameter λ on the RMSECV values: (a) API of pharmaceutical tablets, (b) total alkaloid content and (c) total sugar content in tobacco leaves |
通过与PDS方法比较, 考察了NTRCT方法的应用效果及标准样品的数量因素对这两种方法模型转移效果的影响。
对于药物数据来说, 如果用主机光谱建立的PLS模型直接预测子机光谱样本得到的RMSEP为8.3 mg, 是主机RMSEP的2.4倍, 对应的RPD值为2.65, 预测误差较大, 因此不能满足模型转移实际应用的需要。 PDS和NTRCT方法都能有效降低转移模型的预测误差, 且两者的RPD值均大于3.0(表1), 其中NTRCT的效果接近或好于PDS。
| 表1 药物活性成分的模型转移结果 Table 1 Model transfer results of the API content (mg) in pharmaceutical tablets |
烟叶中的总植物碱和总糖含量是评价烟叶质量的重要化学指标, 因此, 其快速准确的测定是非常重要的。 若用主机样本光谱建立的校正模型直接预测子机光谱样本的总植物碱和总糖含量, 得到的 RMSEP分别为0.49%和1.92%, 对应的RPD值为1.30和3.39(表2和表3)。 表2和表3的结果表明PDS和NTRCT方法都能有效提高转移模型的预测能力, 所得RPD值都大于3.0, 其中NTRCT的效果远远好于PDS方法。 使用15个标准样品时, NTRCT方法模型转移后的RPD值分别增加到6.68和7.84。
| 表2 烟叶中总植物碱的模型转移结果 Table 2 Model transfer results of the total alkaloid content (%) in tobacco leaves |
| 表3 烟叶中总糖的模型转移结果 Table 3 Model transfer results of the total sugar content (%) in tobacco leaves |
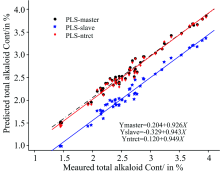
图4比较了使用120个主机样本光谱建立的烟叶中总植物碱含量的校正模型分别对49个样本的主机和子机光谱进行直接预测以及经过NTRCT算法转移后的预测相关分析结果。 我们可以看出, 经过NTRCT算法转移后子机样本光谱的预测精度有显著的提高, 参考值与预测值相关曲线的截距变小(0.120), 对应的斜率更接近于1.0, 与使用主机校正模型直接预测主机样本光谱所得结果较为一致, 这说明了该方法的有效性。
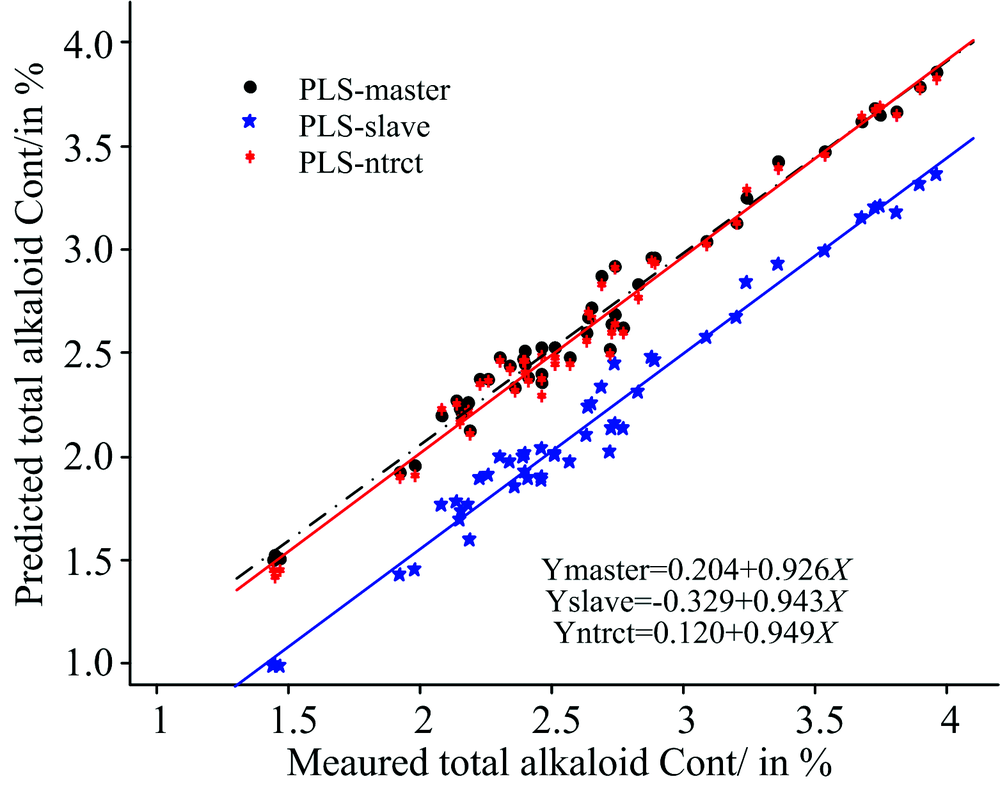 | 图4 模型转移前后烟叶测试样品中总植物碱的预测值与参考值的比较Fig.4 Comparison of predicted and reference values of total alkaloids of tobacco leaf samples before and after model transfer |
随着标准样品数的增加, PDS方法使得药物活性成分主机与子机样品光谱转移模型的预测能力变差(表1); 对烟叶数据来说, 标准样品数量的增加使PDS模型转移方法的预测效果变得更好, 而对NTRCT模型转移方法的预测影响不大。 这说明选择10个或者15个标准样品用于模型转移已足够求得合理的子机模型参数。
基于参数校正的近红外光谱模型转移方法通过使用标准样本集光谱, 正则化主机与子机的光谱模型系数, 训练、 优化参数λ, 实现子机光谱的模型参数校正, 达到较准确的模型转移目的。 该方法分别成功应用于药物活性成分和烟叶中总植物碱与总糖含量的模型转移和预测分析, 使得子机光谱样本的RMSEP明显降低, 且模型预测的RPD值均大于3, 模型预测效果良好。 该方法为具有标准样本模型转移提供了一种思路, 有利于实现近红外模型的共享, 减少重复建立模型的工作量, 从而推动近红外光谱技术网络化的发展。 该方法不适合用于无标准样本的模型转移。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|