{kind=link}
{kind=link}
{kind=link}
{kind=link}
铬掺杂硅团簇的结构、 稳定性和光电子谱性质研究
[林琳1  , 杨桔材
, 杨桔材2, * , 迎春1 , 李继军1 , 赵二俊1 ]
, 杨桔材, 迎春|
|


作者简介: 林 琳, 女, 1974年生, 内蒙古工业大学理学院物理系副教授 e-mail: linlin@imut.edu.cn
采用CCSD(T)/aug-cc-pVTZ-DK//MP2/6-31G(2df, p)和B3LYP/ aug-cc-pVTZ基组研究了小尺寸团簇CrSi n (n=3~9)及其阴离子的结构、 稳定性以及光电子谱。 结果表明: 中性及其阴离子的基态结构是外嵌结构。 由计算得出的解离能可知, 在 n<5时, CrSi n中性结构的稳定性弱于其阴离子结构。 在 n≥5时, CrSi n中性结构中, CrSi5和CrSi8结构的稳定性强于其相邻团簇; CrSi n阴离子结构中, CrSi4和CrSi7结构的稳定性弱于其相邻团簇。 计算得出的CrSi n垂直电子解离能分别为: CrSi3 (2.26 eV), CrSi4 (3.21 eV), CrSi5 (2.72 eV), CrSi6 (3.54 eV), CrSi7 (2.45 eV), CrSi8 (2.71 eV)和CrSi9 (2.95 eV)。 除CrSi4以外, 其他CrSi n结构的垂直电子解离能数值与实验值很好符合, 平均绝对误差仅为0.073 eV。 计算得出的CrSi n绝热电子亲和能分别为: CrSi3 (2.07 eV), CrSi4 (1.95 eV), CrSi5 (2.4 eV), CrSi6 (2.32 eV), CrSi7 (2.38 eV), CrSi8 (2.67 eV)和CrSi9 (2.63 eV)。 除CrSi6以外, 其他CrSi n结构的绝热电子亲和能与实验值很好符合, 平均绝对误差仅为0.09 eV。 此外, 在PBE1PBE/6-31G(2df, p)水平下模拟了CrSi n (n=3~9)阴离子基态结构的光电子光谱, 并与报道的实验结果相比较, 可以得出该研究得到的基态结构是可靠的。
The structures, stability and spectroscopic property of Chromium doped small silicon clusters CrSi n (n=3~9) and their anions are systematically investigated using CCSD(T)/aug-cc-pVTZ-DK//MP2/6-31G(2df, p) and the B3LYP/ aug-cc-pVTZ basis set. The results show that the ground-state structures of neutral CrSi n (n=3~9) and their anion are all exohedral structures. According to the calculated dissociation energies, it shows that when n<5, the neutral CrSi n are less stable than their anion. And when n≥5, the CrSi5 and CrSi8 of neutral CrSi n are more stable than their neighboring clusters; the CrSi4 and CrSi7 of anionic CrSi n are less stable than their neighboring clusters. The VDEs of CrSi n are predicted to be 2.26 eV for CrSi3, 3.21 eV for CrSi4, 2.72 eV for CrSi5, 3.54 eV for CrSi6, 2.45 eV for CrSi7, 2.71 eV for CrSi8 and 2.95 eV for CrSi9. They are in excellent agreement with experimental data except for CrSi4, the average absolute deviations from experimental data are only 0.073 eV. The AEAs of CrSi n are evaluated to be 2.07 eV for CrSi3, 1.95 eV for CrSi4, 2.4 eV for CrSi5, 2.32 eV for CrSi6, 2.38 eV for CrSi7, 2.67 eV for CrSi8, and 2.63 eV for CrSi9. Except for CrSi6, they are in excellent agreement with experimental data. The average absolute deviations from experimental data are only 0.09 eV. Besides, the photoelectron spectra (PES) of ground-state structures of anionic CrSi n (n=3~9) are simulated at the PBE1PBE/6-31G(2df, p) level, and compared to the corresponding experiment data, it is concluded that the ground-state structures obtained in this paper are reliable.
由于硅在半导体工业中的重要性激发了人们对小硅团簇的研究兴趣[1, 2, 3, 4, 5]。 纯硅因有未配对电子导致不稳定。 过渡金属有富裕电子, 若掺入硅团簇中, 可以增加硅团簇的稳定性。 在近几十年来, 许多研究者从理论和实验对过渡金属掺杂硅团簇进行研究[6, 7, 8, 9, 10, 11, 12, 13, 14]。 Beck[6, 7]最早在实验上研究了金属掺杂硅团簇, 报道了
铬掺杂硅团簇也研究报道过。 如Kong等[19]基于密度泛函理论和阴离子光电子谱研究
本文所有计算均在高斯09[22]软件包下进行的。 计算中性CrSin (n=3~9)及阴离子团簇分为以下三步骤。 首先, 采用B3LYP/aug-cc-pVTZ和 MP2/6-31G(d) 理论对结构进行最初的优化和频率的计算; 第二步, 用微扰MP2/6-31G(2df, p)理论进行结构优化; 最后采用CCSD(T)/ang-cc-pVTZ-DK计算上一步结构的单点能。 为能找到能量最低的稳定结构, 对很多的异构体进行计算, 在这篇文章里, 考虑了四种构型, 这几种构型在文献[23, 24]提到过, 这里就不再重述。 另外, 对每个构型, 考虑了自旋多重态, 中性团簇计算了一、 三、 五和七态而相应的阴离子团簇考虑了二、 四和六态。
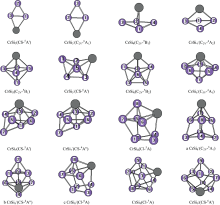
在MP2/6-31G(2df, p)框架下, 对中性的CrSin (n=3~9)和它们的阴离子进行了优化, 如图1所示。 从这些计算结果可以看出, 对中性团簇基态稳定结构是五态的, 而阴离子团簇, 最稳定的结构除n=4, 6, 7是四态的, 其余团簇是六态最稳定。 而且, 除CrSi7基态结构是在异构体GrSi6吸附硅原子得到外, 其余团簇不论是中性CrSin (n=3~9)还是它的阴离子团簇, 最稳定的基态结构几乎都是从纯硅团簇Sin或Sin+1吸附一铬原子或替换一硅原子为铬原子而来得到的。 另外在本文中列出能量相近CrSi8阴离子3种的同分异构体, a, b和c结构。 从能量的角度分析, a结构能量最低, b和c结构能量比a分别高出0.000 6和0.002 eV。 从模拟的光电子谱结构b和c与实验相似程度很大, 但从表1中的垂直电子解离能和亲和能结构c与实验值吻合的很好。 因此在本文结构c定义为阴离子的稳定结构。
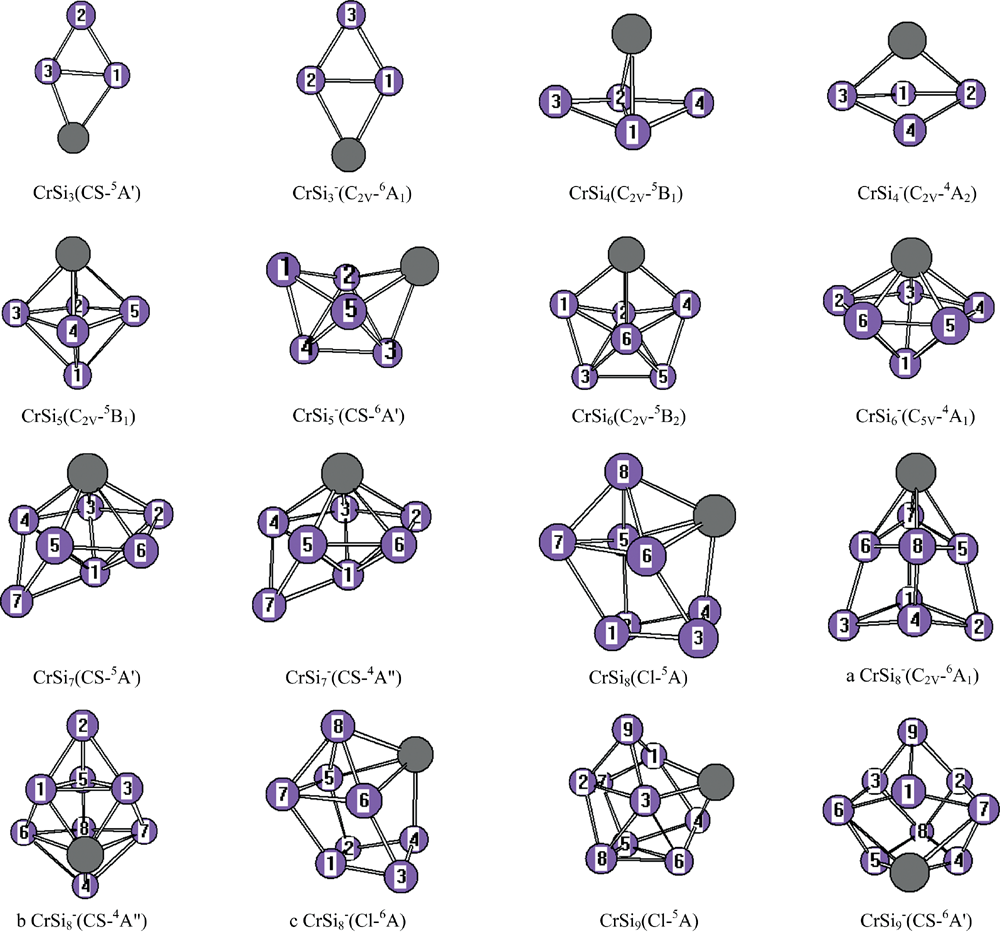 | 图1 CrSin (n=3~9)与其阴离子团簇基于MP2/6-31G(2df, p)优化的结构Fig.1 The isomers of CrSin (n=3~9)and their anions are obtained at the MP2/6-31G(2df, p) level. Only silicon atoms are numbered |
| 表1 CrSin (n=3~9)团簇的绝热电子亲和能和垂直电子解离能 Table 1 Adiabatic electron affinity (AEA) and vertical detachment energy (VDE) for CrSin clustersa |
在耦合簇理论下, 计算了垂直解离能( VDEs)和绝热电子亲和能( AEAs), 见表1。 从表中可以看出, 在CCSD(T)/ang-cc-pvTZ-DK框架下, 计算出的VDEs和AEAs与文献[19]符合得很好, 平均绝对误差分别为0.073和0.09 eV, 这说明我们所选的方法是可信的, 所得到的结构是可靠的。 表中也可以看出, CrSi6的绝热电子亲和能相差比较大, 这是由于CrSi6阴离子电子光谱有很长并圆润的尾巴, 这样就很难准确地测出它的亲和能。
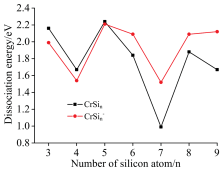
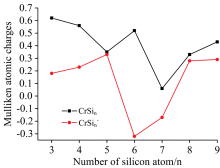
解离能也被详细地计算, 列在表2并绘制在图2。 CrSin团簇的稳定性可从解离能得到, 高的解离能暗示该团簇相对其他团簇较稳定。 从表2以及图2, 可以得到(1)中性团簇比相应的阴离子团簇稳定性要弱, 除n=3, 4和n=5外。 这一点很容易理解, 从密立根电荷如图3所示, 当n≤ 5中性团簇共价性要强于阴离子团簇的共价性, 而其他团簇则是阴离子的共价性强于中性团簇的共价性。 (2)中性CrSi5和CrSi8稳定性高于其他相邻团簇。 (3)中性CrSi4和CrSi7稳定性低于其他相邻团簇。
 | 图2 随硅原子数增加CrSin(n=3~9)中性及阴离子团簇的解离能Fig.2 Dissociation energy (eV) for the reaction CrSin→ Cr+Sin and CrS |
 | 图3 CrSin (n=3~9)中性及其阴离子团簇的自然电荷布局Fig.3 Mulliken atomic charges on Cr atom for CrSin (n=3~9) clusters and their anions |
| 表2 CrSin (n=3~9)中性及其阴离子团簇的解离能(eV) Table 2 Dissociation energies (DEs) for the neutral CrSin and their anionsa |
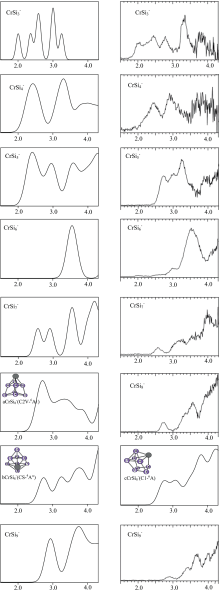
根据广义的库普曼定理, 模拟基态阴离子
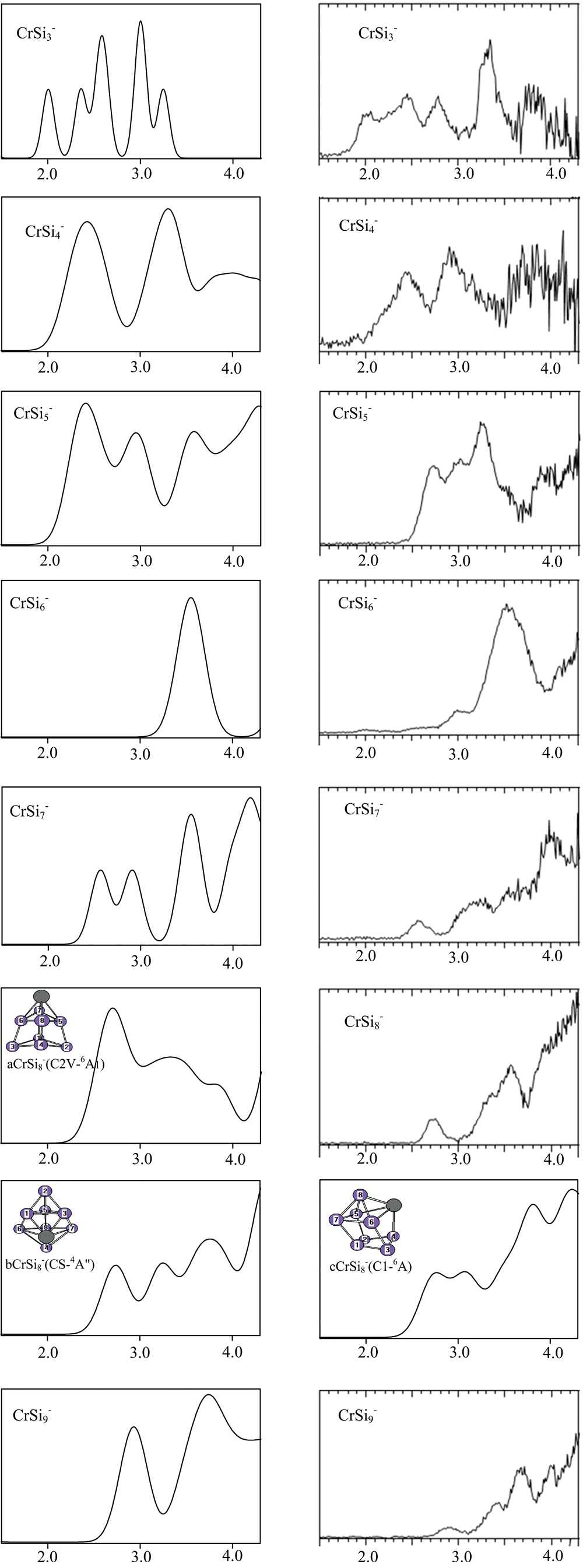 | 图4 在PBEPBE1/6-31G(2df, p)下, 模拟的CrS |
在CCSD(T)/aug-cc-pVTZ-DK//MP2/6-31G(2df, p)理论下, 研究了铬掺杂小尺寸硅团簇结构的稳定性、 能量等性质。 计算结果表明: (1) CCSD(T)框架下, 对过渡金属掺杂硅团簇得到的基态结构和能量是可靠的; (2)除CrSi4外计算得到的垂直电子解离能与文献[19]结果吻合的很好, CCSD(T)计算的理论值与之前测量的光电子谱一致, 说明得到的阴离子团簇基态结构是可靠的; (3)除CrSi6外, 计算得到的绝热电子亲和能与实验数值也符合的很好, 这说明对其他得到的团簇基态结构是可靠的; (4)从解离能可推断出n=3~5中性团簇比相应的阴离子团簇稳定性差, n=5和8时中性团簇比相邻的其他团簇稳定性要好, 阴离子团簇在n=4和7时稳定性比其他相邻团簇弱。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|