
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
高盐样品中锂的电感耦合等离子体发射光谱法(ICP-OES)测定研究
[韩涛1  , 余晓平
, 余晓平1, * , 郭亚飞1 , 李明礼2 , 多吉2 , 邓天龙1 ]
, 余晓平, 郭亚飞|
|


作者简介: 韩 涛, 1993年生, 天津科技大学化工与材料学院硕士研究生 e-mail: 2534691988@qq.com
锂是重要的战略金属和新能源材料, 其开发利用受到全球的高度关注。 在高盐卤水特别是盐湖卤水和地下卤水中富含巨量的锂资源, 在对这些资源进行锂的开发利用过程中, 需对锂的含量进行准确测定, 然而卤水中共存的高浓度Na+, K+, Ca2+和Mg2+会对微量锂的准确测定产生严重的干扰。 电感耦合等离子体发射光谱法(ICP-OES)具有线性范围宽和多元素同步分析能力, 针对卤水中锂的快速准确测定, 详细开展了高盐样品中锂的ICP-OES分析方法研究。 结果表明, 锂在610.364 nm处具有较高的信噪比, 且Na+, K+, Ca2+, Mg2+和Ar不会对锂的测定产生显著的谱线干扰。 然而, 样品中大量共存的Na+, K+和Mg2+会对锂的测定产生严重的基体正干扰, 而Ca2+产生负干扰。 尽管内标法在消除基体干扰方面具有广泛的应用, 但传统的以钇和钪为内标元素的内标法不能有效解决该问题。 此外, 针对标准加入法操作繁琐、 不适合批量样品分析问题, 以及基体匹配法需多离子匹配, 且不适合样品基体组成变化的批量样品分析等问题, 考察了采用单一组分进行复杂基体匹配的可行性。 由于NaCl广泛存在于卤水中, 且对锂的测定具有显著的增敏效应, 通过系列研究发现, 通过同时向样品和标准溶液中加入10 g·L-1的NaCl, 成功解决了总量不超过40 g·L-1的NaCl, KCl和MgCl2所产生的干扰。 尽管采用该法或沉淀预分离方式均不能消除Ca2+产生的负干扰, 但当样品中Ca2+含量不高于1.8 g·L-1时, 对测定不产生显著的影响。 通过采用该方法对三种不同基体组成的卤水样品进行加标回收测定, 其回收率在96.60%104.20%范围内。 此外, 通过与电感耦合等离子体质谱法(ICP-MS)测定结果进行对比, 充分论证了该法的准确性和可靠性(相对误差±3.66%)。 该法仅以单一的NaCl进行复杂基体匹配, 不仅简化了操作, 还实现了基体组成变化的批量样品分析, 因而在卤水中锂的快速准确测定及锂资源开发利用方面具有重要意义。
Lithium is an important strategic metal and new energy material, and its development and utilization has attracted extensive attention worldwide. There are huge amounts of lithium resources in high salinity brines especially in salt lake brines and underground brines. It is necessary to accurately determine lithium content during the development of these resources. However, high concentrations of Na+, K+, Ca2+ and Mg2+ in brines will result in serious interference with the determination of trace lithium. Inductively coupled plasma optical emission spectrometry (ICP-OES) has the ability of multi-element simultaneous analysis with wide linear range. In order to realize swift and accurate determination of lithium in brines, the analysis of lithium in high salinity samples by ICP-OES was carried out in detail in this work. Results showed that lithium has a higher signal to noise ratio at 610.364 nm, where no obvious spectral line interferences of Na+, K+, Ca2+, Mg2+ and Ar were presented for the determination of lithium. However, large amount of Na+, K+ and Mg2+ coexisting in the samples would lead to positive matrix interferences, while the Ca2+ caused negative interference. The internal standard method widely used to eliminate matrix interferences could not effectively solve this problem using whether yttrium or scandium as an internal standard element. Because the standard addition method involves complex operations and is not suitable for batch sample analysis, while the matrix matching method must match these interfering components with corresponding ions and thus is not suitable for the analysis of batch samples with different matrix compositions, the feasibility to match complex matrix with a single component was investigated in this work. Since NaCl widely exists in brines and has obviously sensitizing effect on lithium determination, we found after a series of studies that when the total amounts of NaCl, KCl, MgCl2 in samples were no more than 40 g·L-1, the interferences of these coexisting ions could be successfully solved by adding 10 g·L-1 NaCl into both the samples and the standard solutions. Although the negative interference of Ca2+ could not be effectively solved by this method or pre-separation by precipitation, it did not lead to obvious impact on the determination when its concentration was no more than 1.8 g·L-1. When this method was used for the determination of three kinds of spiked samples with different matrices, the recoveries were within the range of 96.60%~104.20%. Meanwhile, the accuracy and reliability of this developed method were also demonstrated by the comparison with these obtained by the inductively coupled plasma mass spectrometry (ICP-MS) method (The relative errors varied within ±3.66%). Only single NaCl was used in this proposed method to match different coexisting ions, by which it not only simplified the operation, but also realized the batch analysis of samples with different matrix compositions. Therefore, it has important significance for the swift and accurate determination of lithium in brines as well as for the development and utilization of lithium resources in brines.
锂是最轻的金属元素, 具有比热高、 电导性好等特点, 被广泛应用于锂电池、 航空航天等领域[1]。 我国具有丰富的锂资源, 在青海察尔汗和西藏扎布耶等盐湖卤水中, 以及在四川、 山东等地的地下卤水均富含大量的液体锂矿资源[2, 3]。 在对富锂卤水进行锂的开发利用过程中, 需对其含量进行准确测定。 但由于卤水基体组成复杂, 特别是含有大量与锂共存的钠、 钾、 钙、 镁等离子, 给锂的准确测定带来严重的影响[4]。 因此, 开发一种快速准确的高盐卤水中锂的分析测定方法, 对于锂资源的勘探及开发利用等具有重要的意义。
目前, 锂的测定主要采用仪器分析方法, 如电感耦合等离子体质谱法(ICP-MS)[5]、 离子色谱法(IC)[6]、 光谱法等。 其中, 光谱法包括X射线荧光光谱法(XRF)[7, 8]、 原子发射光谱法(AES)[9, 10]、 原子吸收光谱法(AAS)[11] 等。 例如, 张俊文等[5]采用ICP-MS测定了地质样品中锂的同位素, 方法精度优于8‰ ; Zawisza等[7]采用X射线荧光光谱法间接测定了锂的含量, 其分析值与标准值基本一致; 周会等采用火焰原子吸收光谱法(FAAS)测定了铝锂合金中的锂, 其回收率在95.91%99.98%范围内。 值得注意的是, 尽管ICP-MS具有较高的分析灵敏度, 但由于ICP-MS雾化器锥口极小, 高浓度基体会导致雾化器和中心管堵塞等一系列问题。 此外, 尽管AAS可以对高盐基体样品进行测定, 但其不具有多元素同步分析能力。 在对锂资源进行开发利用过程中, 通常需要对样品中不同组分进行测定, 这使得AAS在批量样品测定中的使用受到限制。
AES具有多元素同步分析能力, 其中由于电感耦合等离子体发射光谱法(ICP-OES)采用ICP作为激发光源, 因而具有灵敏度高、 稳定性好、 线性范围广等优势, 在锂的测定方面具有广泛的应用[12, 13]。 例如, Krachler等[12]采用ICP-OES同步测定了放射性样品中的锂、 钠、 钾, 其锂的检出限达到0.15 μ g· L-1。 值得注意的是, 由于卤水中锂的含量较低, 通常处于mg· L-1水平, 而其中共存的Na+, K+, Ca2+和Mg2+等离子的浓度常达到g· L-1甚至接近饱和。 高浓度基体带来的基体效应、 光谱干扰等, 会对锂的准确测定造成严重的影响。 为实现复杂基体中微量组分的准确测定, 常采用标准加入法、 内标法、 基体匹配法等来消除干扰。 标准加入法操作繁琐, 不适用于大批量样品的快速定量分析。 相对于标准加入法而言, 内标法和基体匹配法具有样品处理简单、 试剂消耗量少、 易于批量样品测定等优势。 例如, 袁红战等[14]采用ICP-OES对卤水中高含量锂进行测定时, 采用基体匹配法消除干扰, 尽管其回收率在97.4%101.5%范围内, 但其基体匹配时采用多离子匹配, 操作过程较为繁琐, 且不适合于基体组成变化的批量样品测定。 通常, 基体匹配法处理时, 需要在标准溶液中加入与待测样品组分相同且含量相近的基体元素, 若样品基体组成复杂, 则该法的应用十分繁琐。
目前, 与锂分析相关的研究主要集中在对单离子基体或不涉及高Na+, K+, Ca2+和Mg2+样品中锂的分析。 由于ICP-OES具有灵敏度高、 多元素同测等特征, 因此本文针对高盐卤水中锂的准确测定问题, 以ICP-OES为工具, 开展卤水中锂的分析方法研究, 为卤水中锂的开发利用奠定基础。
主要仪器: Prodigy-H电感耦合等离子体发射光谱仪(ICP-OES, 美国Leeman, 仪器参数见表1); 电子分析天平(± 0.1 mg, 美国OHAUS); 电感耦合等离子体质谱仪(ICP-MS, 美国Thermo Fisher)。
| 表1 ICP-OES工作参数 Table 1 Working parameters of ICP-OES |
主要试剂: 1 000 mg· L-1锂、 钇、 钪标准储备液(国家钢铁材料测试中心); 实验用药品NaCl、 KCl、 无水CaCl2、 无水MgCl2、 硝酸均为优级纯(国药集团化学试剂有限公司)。 实验用卤水为基体组成不同的三种真实卤水样品(盐湖卤水、 地下卤水、 地热水), 其组成见表2。
| 表2 卤水样品组成 Table 2 Composition of brine samples |
1.2.1 标准曲线绘制
将1 000 mg· L-1锂标准储备液用3%硝酸稀释于同一规格容量瓶中, 配制浓度依次为0, 20, 40, 60, 80, 100, 200, 300, 400和500 mg· L-1的锂标准系列溶液。 在表1条件下, 采用ICP-OES进行测定, 根据测定结果评估ICP-OES测定锂的线性范围及线性相关性。
1.2.2 水样分析
根据锂标准曲线线性范围, 对水样进行适当稀释(地下卤水稀释10倍, 盐湖卤水稀释2倍, 地热水不稀释), 并用浓硝酸调节溶液硝酸浓度为3%。 在与标准曲线测定相同的条件下进行测定, 并采用标准曲线法进行回收率计算, 根据回收率评估基体的干扰。 其计算公式如式(1)
式中: wLi为Li+测定, 回收率(%); c'Li为Li+测定浓度(mg· L-1); cLi为Li+真实浓度(mg· L-1)。
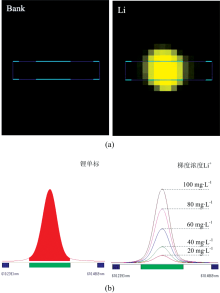
在3%的硝酸介质中, 0500 mg· L-1浓度范围内锂的标准曲线都具有很好的线性相关性(r> 0.999 5), 检出限(DL)仅为0.007 8 mg· L-1。 然而, 当直接采用仅含3%硝酸介质的标准曲线对表2中三种样品进行测定时, 锂的回收率分别高达145.41%, 141.76%和119.91%。 由于卤水中含有高浓度的无机盐, 其误差可能来源于共存离子导致的光谱或基体干扰。 尽管锂的特征谱线众多, 但由于谱线信噪比及其他元素谱线的干扰等问题, 经实验发现, 在所考察的浓度范围内, 锂在610.364 nm处信噪比较高, 且Na+, K+, Ca2+, Mg2+和Ar在该谱线附近不会产生光谱干扰(表3、 图1)。 因此, 对卤水样品进行ICP-OES测定的误差主要来源于基体干扰。 例如, 由于样品组成及表面张力等性质与标准溶液不同, 导致进样速率、 进样量、 雾化效率、 在等离子体中激发形成的原子或离子数量等存在差异, 从而造成测定误差。
| 表3 具有较高信噪比的锂、 钠、 钾、 钙、 镁元素 及氩气的分析谱线 Table 3 Spectral lines of lithium, sodium, potassium, calcium, magnesium, argon with high signal-to-noise ratio |
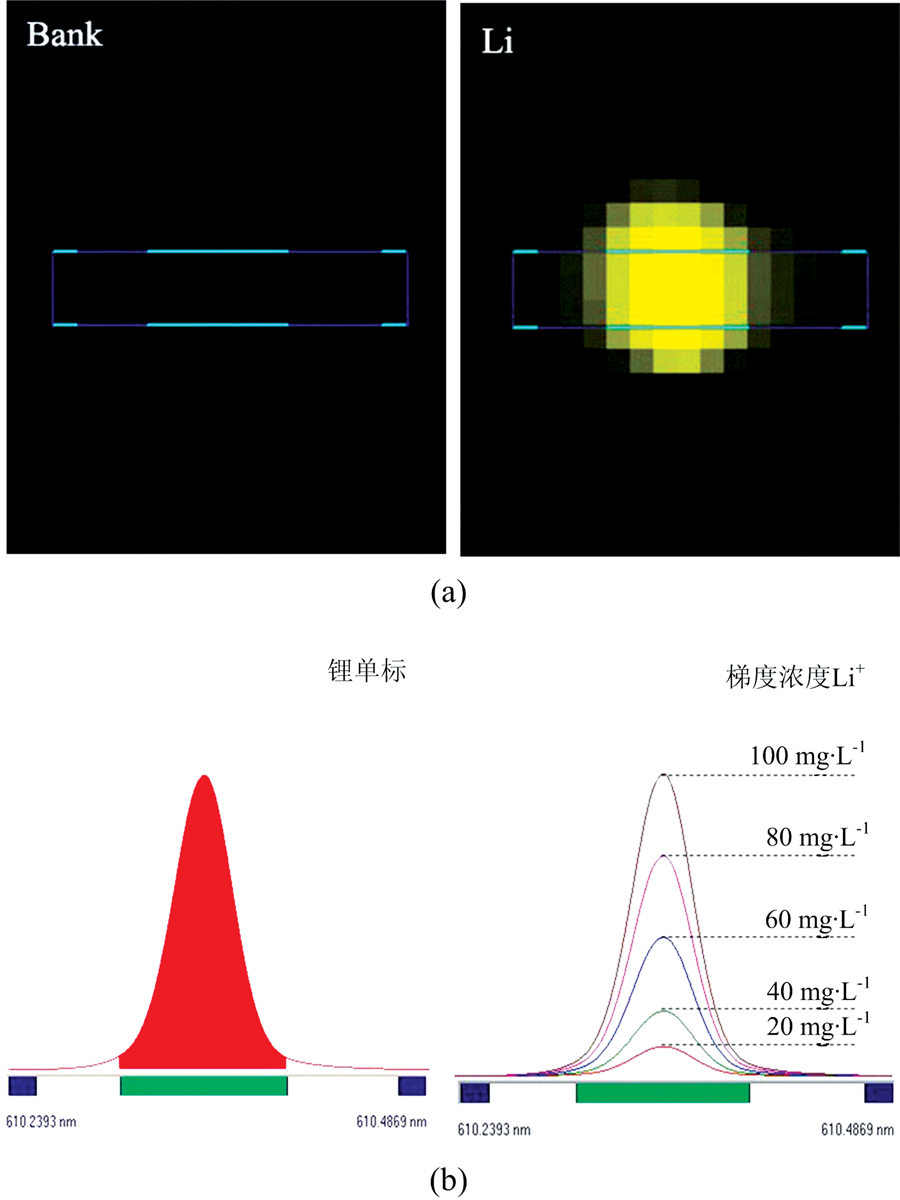 | 图1 锂的谱线干扰(a)及谱线处理(b)Fig.1 Spectral line interference of lithium (a) and its process (b) |
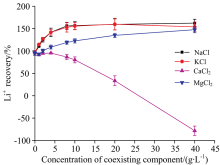
为揭示不同共存离子(Na+, K+, Ca2+, Mg2+)对锂测定的影响, 分别配制系列含不同浓度(040 g· L-1)NaCl, KCl, CaCl2和MgCl2的锂溶液, 并控制锂的浓度均为20 mg· L-1。 采用3%硝酸介质为基体的标准系列溶液进行定量, 结果见图2。 由图可知, 在一定浓度范围内, 四种组分均对锂的测定产生严重的干扰, 且组分含量越高, 其干扰程度也越大。 通常, 当样品中含有大量易电离元素时, 将导致等离子火炬中的自由离子和电子过于丰富, 从而在一定程度上抑制待测组分的电离, 导致测定结果比实际含量偏低。 但从实际测定结果来看, 尽管Ca2+对测定结果表现为强烈的抑制作用, 但Na+, K+和Mg2+均表现为增敏作用, 其中Na+和K+的增敏效应尤为严重。 其主要原因在于, 不同观察区域基体效应的性质有所不同, 钠、 钾、 镁对锂测定的增感效应符合低观察区域基体元素碰撞激发的干扰机理, 故表现为增敏作用。 而抑制一般出现于正常分析区, 且在同一观测高度, 钙的抑制幅度远大于镁。 当所考察的共存组分浓度超过15 g· L-1时, KCl, NaCl和MgCl2对锂测定的影响逐渐趋于稳定, 而CaCl2的抑制作用逐渐增强。 当共存组分浓度超过60 g· L-1时, ICP-OES火焰极不稳定, 甚至熄灭, 因此未进行更高浓度的实验。
 | 图2 共存离子对锂测定的影响Fig.2 Effects of co-existing ions on lithium determination |
2.3.1 基于内标法的共存离子干扰消除
内标法是一种常用的消除基体干扰的方法, 在光谱定量分析中有广泛的应用。 由于样品中发生的一切改变在内标中同样存在, 即样品与内标在样品引入或等离子体中所发生的变化是一致的。 在测量过程中内标信号增加或降低的系数与样品相同, 因此通过计算二者强度的比值可以校正基体干扰。 通常, 卤水样品中Na+的含量较高且对锂测定影响最为显著, 因此实验首先考察分别以常用的钇(Y)和钪(Sc)作内标物来消除共存NaCl的干扰。 实验结果见表4。
| 表4 以钇和钪作内标元素消除NaCl基体干扰结果 Table 4 Results on the elimination of NaCl interference by using yttrium and scandium as internal standard elements |
由表4可知, 尽管采用内标法后, 锂回收率比直接采用3% HNO3介质的标准曲线的回收率低, 但均大于130%, 且随NaCl基体浓度的升高, 干扰程度逐渐增大。 其可能原因在于, 锂、 钠、 钾、 钙、 镁为碱金属或碱土金属, 而钇和钪为第IIIB族过渡金属, 其性质更接近于同族的稀土元素。 此外, 钇和钪的最适谱线分别为371.030和361.383 nm, 这与锂的610.364 nm最适波长相差较大, 这均可能造成基体对锂和内标物干扰程度的差异, 最终导致内标法无法消除共存基体的干扰。
2.3.2 基于单基体匹配的共存离子干扰消除
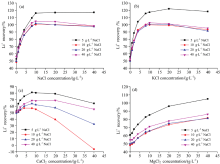
传统的基体匹配法在操作时, 针对不同基体样品的测定, 需分别配制不同的标准系列溶液, 其基体组成需与样品基体一致, 因而操作繁琐、 试剂耗量大, 且不适合批量样品的测定。 因此, 针对卤水中Na+, K+, Ca2+和Mg2+的干扰, 进一步考虑采用单离子基体匹配的可行性。 由于NaCl是卤水中最常见的组分, 因此分别配制了含5, 10, 20和40 g· L-1 NaCl介质的四套标准系列溶液, 并分别以此对含不同浓度NaCl, KCl, CaCl2和MgCl2单一基体的锂(20 mg· L-1)溶液进行测定, 结果见图3(a— d)。
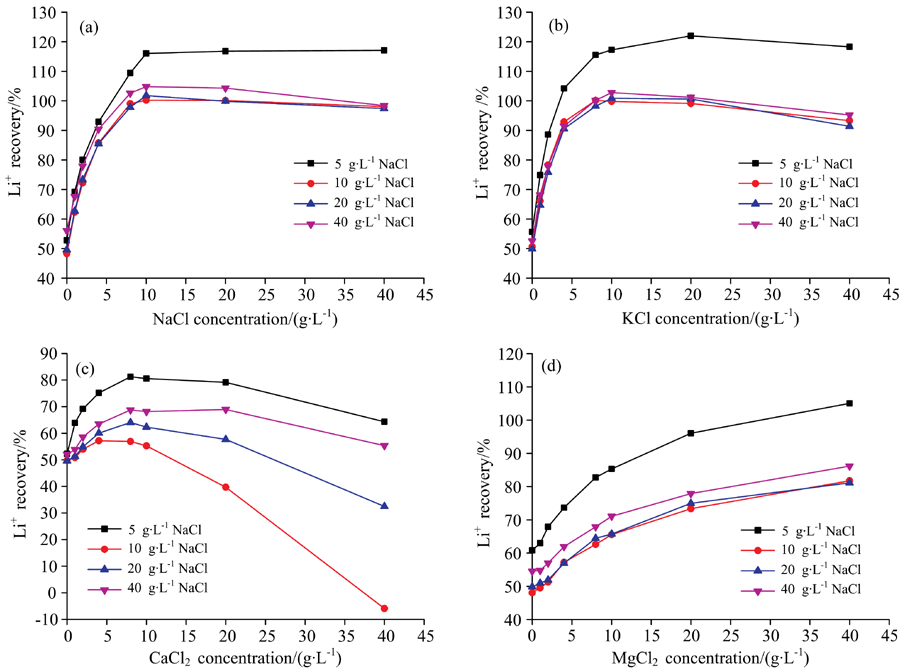 | 图3 采用单一NaCl匹配不同共存离子消除基体干扰的可行性 (a): NaCl; (b): KCl; (c): CaCl2; (d): MgCl2Fig.3 Feasibility for the elimination of matrix interference by matching different coexisting ions with single NaCl (a): NaCl; (b): KCl; (c): CaCl2; (d): MgCl2 |
由图3(a)可知, 当样品中NaCl浓度大于10 g· L-1时, 尽管采用5 g· L-1 NaCl介质的标准系列溶液无法很好地消除干扰, 但采用10, 20和40 g· L-1 NaCl介质的标准系列溶液时, Li+的回收率均在95%105%范围内, 即当标准溶液中NaCl浓度大于10 g· L-1时, 可消除样品中1040 g· L-1 浓度范围内的NaCl干扰。 其结果与图2中结果一致, 即当样品中NaCl超过一定浓度(约10 g· L-1)后, Li+的回收率基本保持不变。 图3(b)中结果与图3(a)中类似, 即通过在标准中添加一定浓度的NaCl可以消除样品中一定浓度范围内KCl的干扰。 值得注意的是, 尽管NaCl对锂的测定产生严重正干扰, 但由图3(c)可知, 仅在标准系列中加入NaCl, 在所考察条件下均不能消除CaCl2产生的负干扰, 即使标准溶液中NaCl浓度高达40 g· L-1。 此外, 针对MgCl2干扰而言, 由图2可知, 相同质量浓度下, NaCl产生的正干扰显著高于MgCl2, 因此在图3(d)中当标准系列溶液中NaCl含量高于10 g· L-1时, 导致在所考察浓度范围的MgCl2基体中锂的回收率均低于100%。 因此, 尽管在标准系列中添加NaCl可很好消除样品中一定浓度范围内的NaCl和KCl干扰, 但不能消除CaCl2和MgCl2的干扰。
由于NaCl对锂的信号产生显著增敏效应, 因此能否在含Ca2+样品中添加适当NaCl以消除CaCl2的信号抑制作用? 同时, 由于NaCl的增敏效应显著高于MgCl2, 能否在含Mg2+样品中添加NaCl来稳定MgCl2的干扰效应? 鉴于此, 分别配制系列不同浓度的含单一CaCl2和MgCl2基体的锂(20 mg· L-1)溶液, 再在其中加入等质量浓度的NaCl(10 g· L-1, 由于样品中NaCl含量超过10 g· L-1后, 其对锂信号的影响基本不变, 故选择该浓度), 并采用3% HNO3介质的锂标准系列溶液进行测定, 结果见图4。
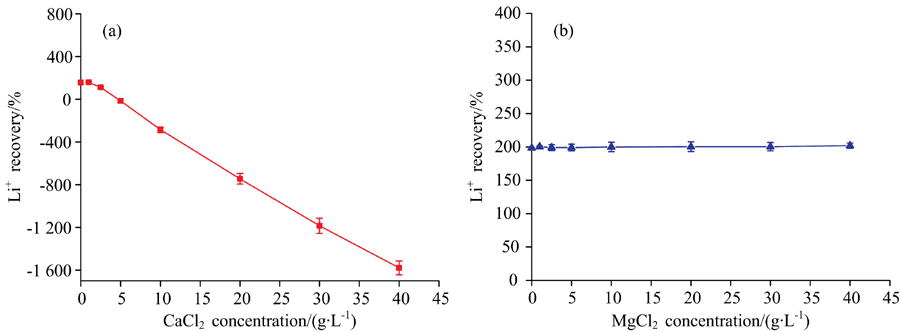 | 图4 样品中添加10 g· L-1 NaCl校正Ca2+和Mg2+干扰的可行性 (a): CaCl2; (b): MgCl2Fig.4 Feasibility for the calibration of Ca2+ and Mg2+ interference by adding 10 g· L-1 NaCl into sample (a): CaCl2; (b): MgCl2 |
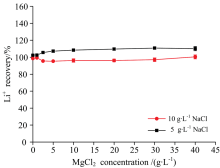
由图4(a)可知, 尽管NaCl能显著增加Li+的信号强度, 但当样品中CaCl2浓度高于5 g· L-1时, 即使在样品中添加10 g· L-1 NaCl也未能改善CaCl2的负干扰。 与此不同, 由图4(b)可知, 尽管在含MgCl2的样品中添加10 g· L-1 NaCl后锂的回收率高达200%, 但在所考察的MgCl2浓度范围内, 锂的回收率基本不变, 即NaCl的强正干扰可湮灭MgCl2的干扰。 因此, 可以通过同时向标准和样品中加入NaCl来消除MgCl2的干扰, 或当样品中存在一定浓度的NaCl时, 可以不考虑MgCl2产生的干扰。 为揭示向样品和标准中同步加入NaCl的浓度, 以及该法适用的MgCl2浓度范围, 进一步以含5和10 g· L-1 NaCl基体的锂标准溶液对含10 g· L-1 NaCl且MgCl2浓度梯度变化的锂(20 mg· L-1)溶液进行测定, 结果见图5。
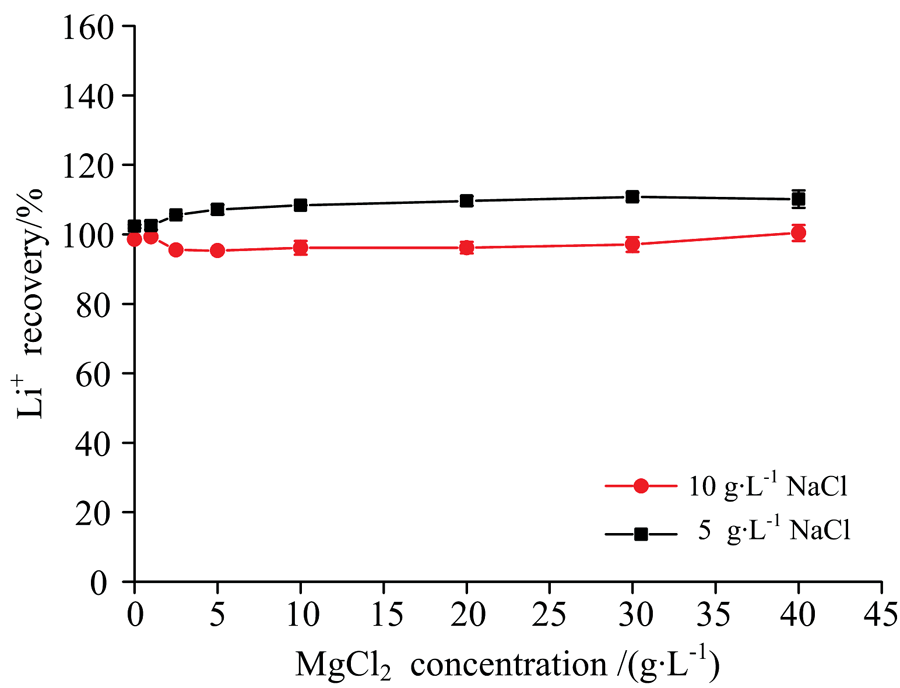 | 图5 标准和样品中同时加入NaCl消除Mg2+干扰Fig.5 Elimination of Mg2+ interference by simultaneously adding NaCl into standard solutions and sample |
由图5可知, 无论采用含5或10 g· L-1 NaCl基体的标准溶液对含MgCl2的样品进行测定, 当同时向样品和标准系列溶液中加入10 g· L-1 NaCl时, 样品中MgCl2含量在040 g· L-1范围内均对锂的测定不产生干扰, 其回收率稳定在95%105%范围内, 实现了含MgCl2基体中锂的准确测定。
2.3.3 共存钙离子干扰消除
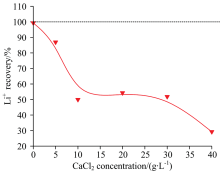
由于无论采用内标法还是改进的基体匹配法均不能消除CaCl2产生的负干扰, 因此考虑采用沉淀预分离方式对Ca2+进行处理。 尽管CaCO3具有极低的溶解度(Ksp=2.8× 10-9), 但在以C
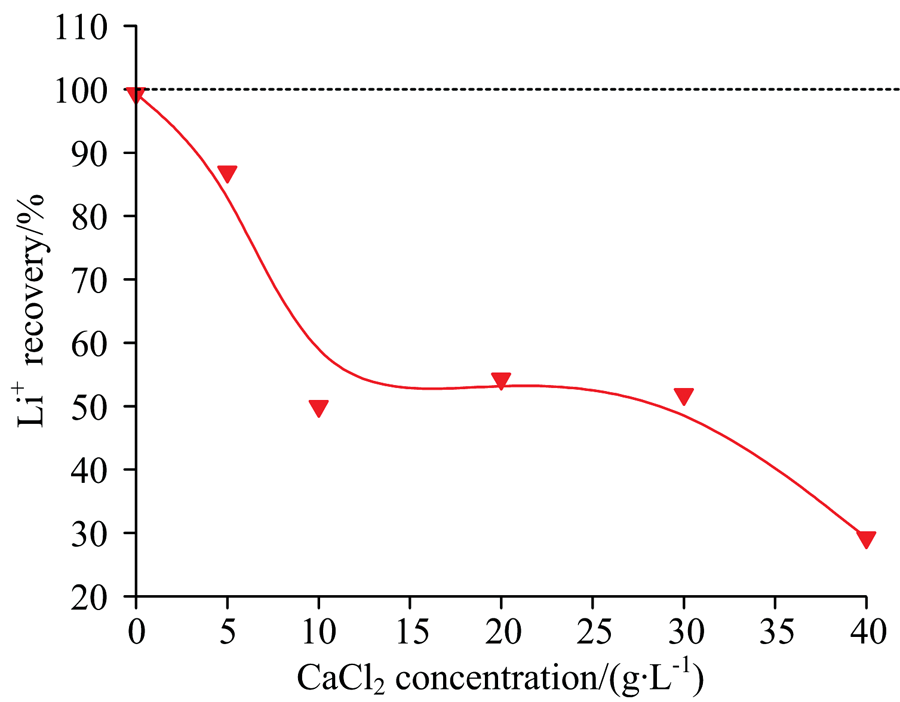 | 图6 C2H2O4沉Ca2+时Li+的夹带或共沉淀损失Fig.6 Loss of Li+ by entrainment or co-precipitation during the precipitation of Ca2+ with C2H2O4 |
由图6可知, Ca2+在约04.5 g· L-1(CaCl2: 012.5 g· L-1)范围内时, 随Ca2+浓度的增加, Li+的回收率急剧降低。 当Ca2+浓度超过1.8 g· L-1(CaCl2: 5 g· L-1)时, Li+的回收率低于90%。 此外, 若溶液中同时含有Ca2+和M
基于本文研究结果, 当样品中Ca2+含量低于1.8 g· L-1, 且NaCl, KCl和MgCl2总量不超过40 g· L-1时, 可以采用改进的基体匹配法消除共存Na+, K+和Mg2+的干扰, 即通过在样品和标准中同时加入10 g· L-1的NaCl消除总量不超过40 g· L-1的NaCl, KCl和MgCl2的干扰。
为验证方法的可靠性, 采用该法对表2中三种水体样品进行测定。 该法在0300 mg· L-1锂范围内都具有很好的线性相关性(r> 0.999 4), 检出限仅为0.008 5 mg· L-1。 在测定时, 根据锂标准曲线线性范围, 对样品进行适当稀释(地下卤水稀释10倍, 盐湖卤水和地热水不稀释)。 此外, 为便于对比, 采用传统的基体匹配法进行Li+的ICP-MS测定(样品在ICP-OES稀释倍数基础上再稀释500倍), 结果见表5。
| 表5 ICP-OES与ICP-MS测定Li+结果对比 Table 5 Comparison between the results of Li+ determination by ICP-OES and ICP-MS |
尽管地下卤水中Ca2+换为CaCl2后, 其含量高于5 g· L-1(约6.76 g· L-1), 但对样品适当稀释后其含量低于方法要求。 由表5可知, 采用本方法后, 三种样品中Li+的回收率在96.60%104.20%范围内, 满足微量组分定量分析要求。 由于ICP-MS具有极高的灵敏度, 且由于采用了传统的基体匹配法, 故可以用表5中ICP-MS测定结果作为卤水中锂含量的参考值。 基于此, 通过对比表5中两种分析方法的相对误差(均在± 3.66%范围内), 进一步验证了本文所建立方法的可靠性。
ICP-OES具有多元素同步分析能力, 但其在对卤水样品中Li+进行测定时, 高浓度的Na+, K+, Ca2+和Mg2+共存离子会对Li+的准确测定产生严重的干扰。 其中, Na+, K+和Mg2+产生正干扰, 而Ca2+产生负干扰。 采用传统的以Y和Sc为内标元素的内标法均不能消除该干扰。 通过改进传统的在标准溶液中加入各种共存离子的基体匹配法, 建立了一种仅添加NaCl来消除共存NaCl, KCl和MgCl2干扰的改进基体匹配法。 该法通过同步向样品和标准系列溶液中加入10 g· L-1的NaCl, 解决了总量不超过40 g· L-1的NaCl, KCl和MgCl2所产生的干扰, 且当样品中Ca2+含量不高于1.8 g· L-1, 对测定不产生显著影响。 本方法仅以NaCl匹配样品中的其他共存离子, 解决了传统方法需要多离子匹配, 以及一个样品需要匹配一套标准溶液所造成的操作繁琐、 不适合批量样品分析等问题, 因而在卤水中锂资源的开发及综合利用方面具有重要意义。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|