

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
仲氢诱导超极化增强核磁共振技术: 从原理到应用
[王忻昌1, 2  , 江文龙
, 江文龙1 , 黄程达1 , 孙惠军1 , 曹晓宇2 , 田中群2 , 陈忠1, 2, * ]
, 江文龙]
|
|


作者简介: 王忻昌, 1987年生, 厦门大学电子科学与技术学院博士后 e-mail: xcwang@xmu.edu.cn
核磁共振(NMR)技术可提供原位、 实时、 高分辨的分子结构和生物组织信息, 虽然已广泛用于复杂分子结构表征和生物成像, 但低灵敏度限制了其进一步应用。 通过将外源性超极化状态核自旋转移至待测分子的原子核, 实现目标分子的超极化从而提高待测物中原子核特定自旋态的布居数差, 可有效提高NMR灵敏度。 然而, 目前主流超极化方法共同的缺点是仪器昂贵、 操作复杂。 仲氢诱导增强的超极化是一种低成本、 操作简单且高效的技术, 可将NMR灵敏度提高三个数量级以上, 正逐渐成为NMR领域的前沿热点。 介绍仲氢诱导增强超极化的基本原理、 实验方法以及在NMR谱学和成像方面的应用。 仲氢的两个氢原子核自旋方向相反, 通过降温和催化剂作用可实现高纯度富集。 富集后的仲氢通过两种方法将其超极化状态转移至底物分子。 第一种方法是在催化剂作用下将仲氢分子加成到底物分子的不饱和基团上, 直接采集氢的NMR信号, 或通过极化转移方法(脉冲序列或磁场循环)将其超极化状态转移至邻近的异核原子核(如13C,15N,19F等)。 第二种方法是仲氢与底物分子在金属配合物进行可逆交换反应, 实现底物分子中杂核原子的超极化。 选择合适的催化剂对灵敏度提升至关重要, 对常用催化剂的类型和结构进行了总结; 极化转移方法对异核灵敏度的提升至关重要, 并归纳了此技术中常用的极化转移方法。 仲氢诱导超极化增强技术对NMR灵敏度的显著提升使得该技术在应用方面已崭露头角。 首先, 仲氢诱导超极化增强技术将NMR谱所需浓度降低到μmol·L-1级别, 可用于表征催化反应中间体结构及监测混合物中低浓度化学物质。 其次, 基于异核的超极化底物分子是良好的NMR生物成像造影剂。 虽然仲氢超极化增强技术具有广泛的应用前景, 然而理性设计并合成超极化率高、 寿命长、 水溶性好的NMR造影剂仍是亟待解决的关键问题。
, JIANG Wen-long
Nuclear Magnetic Resonance (NMR) can provide real-time, in-situ high-resolution molecular and biological information, and has been wildly applied in determining the structures of highly complex molecules and biological imaging. Its low sensitivity, however, hindered its further application. An effective way to increase the sensitivity of NMR is transferring hyperpolarized spin-orders of exogenous particles to the substrates. The main drawbacks of current methods are high cost of equipment and complicated procedures. Parahydrogen-induced polarization (PHIP) is a potential candidate to increase sensitivity of NMR due to its low cost, simple procedure and high polarization efficiency. By transferring the spin-order of parahydrogen into substrate, the sensitivity of NMR can be increased by at least 3 orders. This review will briefly introduce the basic principles of PHIP, physical and chemical procedures involved and its application in chemistry and biological imaging. The two hydrogen atoms in parahydrogen are in opposite spin states, and can be enriched at low temperature with the help of catalyst. Enriched parahydrogen is relatively stable even when returned to room temperature. Spin-order of parahydrogen can be transferred to substrate by two approaches. First, addition of parahydrogen into unsaturated groups in substrates, which can be used directly, or transfer spin-order to adjacent heteronucleus (13C,15N,19F et al). Second, signal amplification by reversible exchange (SABRE), through which parahydrogen and substrates are reversibly coordinated into metal complex and the spin-order is transferred from parahydrogen to substrates. Choosing the right catalysts is crucial for the enhancement of NMR sensitivity, and this manuscript summarized the types of catalyst used in this technique and their structures. Moreover, methods to transfer the hyperpolarized spin-order of parahydrogen, which are important for enhancing the sensitivity of heteronucleus, were also summarized. PHIP has shown great potential in many applications due to its high increase in sensitivity. First, this technique requires much lower sample concentration (μmol·L-1 or even lower) than normal NMR, which makes determination of low concentration species such as reaction intermediate or trace analysis. Second, hyperpolarized substrates are good candidates for NMR imaging contrast agent. Nevertheless, realizinglong-lived imaging contrast agent with high polarization and good solubility in water is still challenging.
核磁共振(nuclear magnetic resonance, NMR)技术是一种原位实时、 高分辨的非侵入式表征手段, 能提供原子化学环境、 偶合、 化学键连接、 原子空间距离和分子尺寸等重要结构信息[1], 已在化学生物分子表征、 医学成像方面得到广泛应用。 虽然NMR技术已发展几十年, 但因其检测灵敏度低, 要求样品浓度较高、 采集时间偏长, 限制了其更广泛的应用。 NMR技术灵敏度低的最根本原因是核自旋态能级差小, 导致高低自旋态能级间粒子数差仅为10-5数量级。 即使提高超导磁场强度(> 10 T)和将射频线圈冷却到超低温(4 K), 对NMR灵敏度的提升仍很有限, 因此大幅提高NMR灵敏度是一项挑战性很强的工作。
为了提高原子核自旋态高低能级的布居数差, 可将外源性粒子的超极化状态传递到目标分子的原子核[2], 使后者达到超极化状态。 目前主流的超极化方法有动态核极化(dynamic nuclear polarization, DNP)、 自旋交换光泵(spin-exchange optical pumping, SEOP)等。 虽然这些方法可有效提高NMR的灵敏度, 但成本高(昂贵的设备费和维护费)且操作步骤繁琐, 对专业技术要求很高。 为此发展低成本、 易操作、 普适性强的超极化NMR灵敏度增强技术具有重大意义[3]。
最近仲氢(p-H2)诱导增强超极化技术成为广泛关注的热点[4], 其具有设备成本低、 原料易得、 操作简便、 极化效率高等优点, 可望在应用领域大范围推广。 p-H2诱导超极化增强技术的原理是将富集后的p-H2(纯度可大于99%)通过催化加氢反应加成到底物分子, 或通过可逆交换反应将p-H2的超极化状态传递到底物分子的杂核原子(13C, 15N, 19F, 31P等)[5]。 理论上此技术可将NMR的灵敏度提高5个数量级, 实际操作中灵敏度提高比例受H2加成效率、 自旋极化传递效率、 可逆交换效率、 周围是否存在顺磁物质等因素影响[6]。
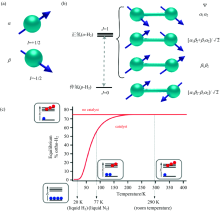
氢原子核自旋量子数I=1/2, 存在+1/2和-1/2自旋态, 对应波函数为α 和β [图1(a)]。 H2存在四个自旋态, 即α α , α β +β α , β β 和α β -β α , 如图1(b)所示。
 | 图1 (a)氢原子的两种自旋态, (b)氢气分子的四种自旋态与转动能级对应, (c)氢气分子中正氢的比例与温度的关系Fig.1 (a) Two spin states of hydrogen atoms, (b) Four spin states of hydrogen molecules and their corresponding rotational states, (c) Relationship between the ratio of o-H2 and temperature |
其中波函数α α , α β +β α 和β β 的三个自旋态为两个氢原子的核自旋方向相同, 称为正氢(o-H2); 波函数α β -β α 的自旋态为两个氢原子的自旋方向相反, 称为p-H2。 o-H2和p-H2不仅自旋态能量不同, 转动态能量也不同, 自旋态能级与其转动能级紧密关联, 这与量子力学中的波函数对称性有关。
H2四种自旋态的布居数由转动基态与转动第一激发态的能级差决定, 室温下此能级差对o-H2和p-H2的玻尔兹曼分布影响很小, 因此四种自旋态的分布比例大致相同, 即o-H2和p-H2的比例约为75%和25%。 由于p-H2处于转动基态, 因此可通过降温提高p-H2的比例。 在77 K时p-H2的比例约为50%, 在20 K时p-H2达到99%以上。 虽然p-H2热力学更稳定, 但由于o-H2和p-H2间转动态和自旋态的对称性不吻合, 因此存在对称性禁阻, o-H2向p-H2转变的动力学过程非常缓慢。 需要加入催化剂加速此转化过程, 通常为铁的氧化物[如FeO(OH)]。 富集后的p-H2回到室温后, 由于p-H2向o-H2转化的动力学过程非常慢, 因此在合适的容器中可长时间储存, 例如在铝制容器中可保存数周[7]。
富集后的高纯度p-H2可通过两种方法将超极化自旋态传递至目标底物分子, 第一种方法是通过催化加氢反应, 即将p-H2直接加成到底物分子的不饱和基团, 称为PHIP(parahydrogen-induced polarization), 第二种方法是p-H2与配合物分子在配位化合物上进行可逆交换反应, 将超极化状态通过自旋-自旋偶合传递至底物分子, 称为SABRE(signal amplification by reversible exchange)。 1.2节简要介绍这两种极化增强方法。
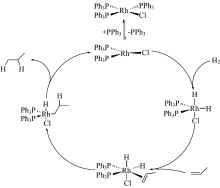
p-H2无核磁信号(I=0), 在PHIP实验中通过化学反应将p-H2加成到底物分子上, 两个超极化氢原子在不同化学环境中呈现增强NMR信号, 或将超极化状态传递到临近的杂核原子。 此过程的关键步骤是将p-H2加成到底物分子, 氢气加成反应是一类常见的化学反应, 通常需要加入金属催化剂, 加成机理为H2和不饱和化合物分别配位到金属中心后, H2加成到不饱和键, 随后产物从金属中心脱掉, 金属催化剂重新进入催化循环(图2)。 目前已有不少催化剂可用于加成反应, 如Wilkinson催化剂, 即三(三苯基膦)氯化铑催化剂[RhCl(PPh3)3], 其加成机理如图2所示。 加氢反应的效率对NMR灵敏度的提高至关重要, 宋艳红等研究了加氢反应条件对最终核磁灵敏度提高的影响[6], 指出增加氢气与反应体系的接触面积、 升高温度和增加氢气压力等可提高加氢反应速率的方法, 可帮助NMR灵敏度的提升。
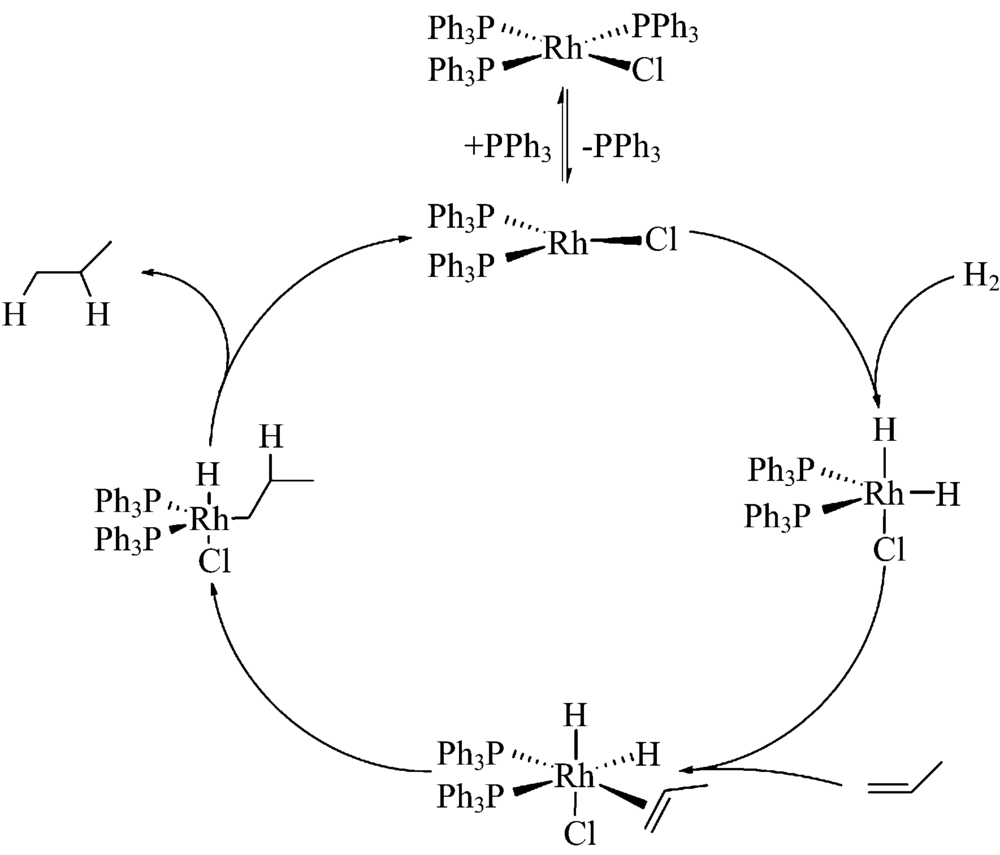 | 图2 Wilkinson催化剂的催化加氢反应机理Fig.2 Mechanism of catalytic hydrogenation of Wilkinson catalyst |
值得注意的是, p-H2加成到底物分子后, 对称性一定会被打破, 即两个氢原子要处于不同化学环境, 否则加成后两个氢原子在核磁谱中不会产生NMR信号, 具体解释可参考文献[8]。 H2加成到底物后, 若形成AX自旋体系, 两个氢原子存在四种自旋态, 即α α , α β , β α 和β β 。 由于四种自旋态能级相差较小, 因此氢原子平均分布在四个自旋态, 存在四种自旋能级跃迁, 在常规氢谱中呈现四条NMR峰[图3(b)]。
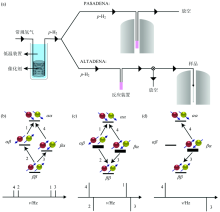
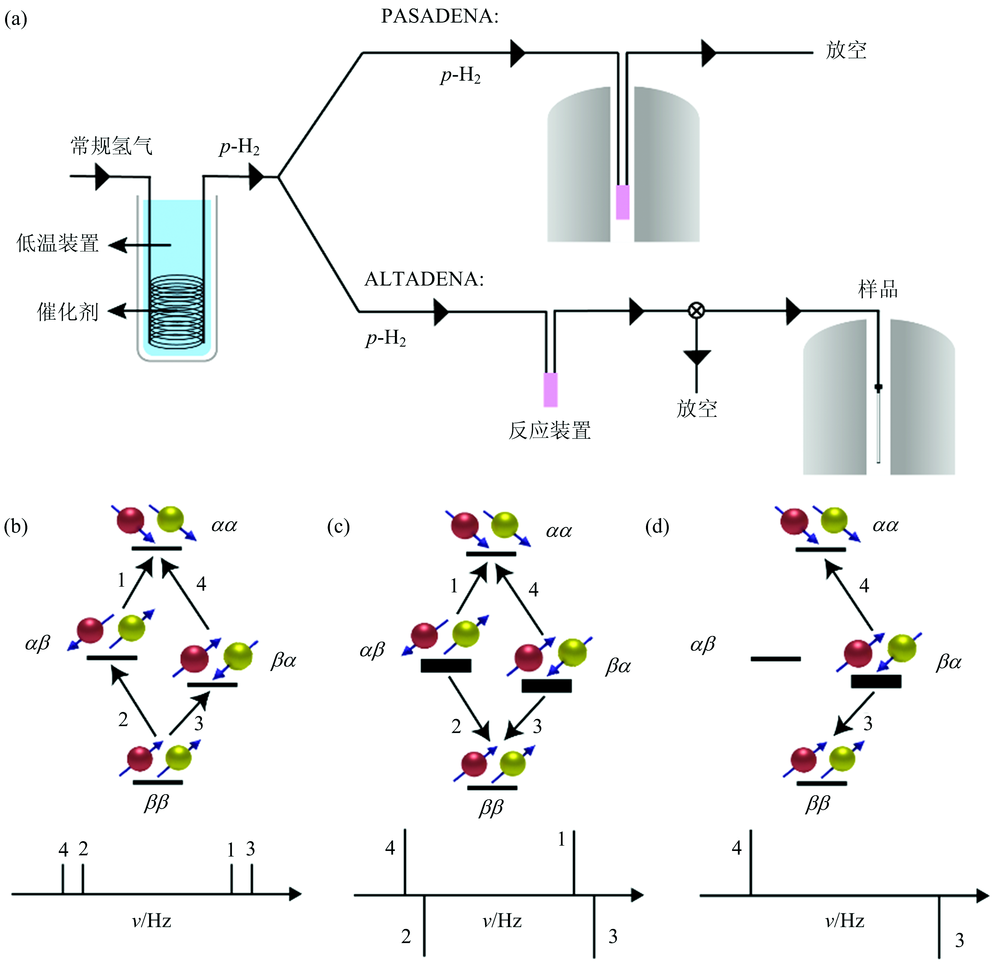 | 图3 (a) p-H2富集装置、 p-H2加成反应的两种不同反应路径; AX自旋体系的能级分布及氢谱(模拟): (b)常规氢谱; (c)PASADENA实验; (d)ALTADENA实验Fig.3 (a) Enrichment of p-H2 and two approaches for hydrogenation of p-H2; Energy states and simulated 1H-NMR spectra of AX spin systems: (b) normal experiment, (c) PASADENA and (d) ALTADENA |
根据p-H2加成反应发生时的磁场强度不同, 有PASANENA (parahydrogen and synthesis allow dramatically enhanced nuclear alignment)和ALTADENA (adiabatic longitudinal transport after dissociation engenders net alignment)两种实验条件: PASANENA在高强度磁场(> 1 T)中进行p-H2加成反应, 而ALTADENA在低强度磁场(地磁场50 T或更低)中进行, 两种策略的反应过程示意图如图3(a)所示。
不同磁场强度下底物的氢原子间自旋-自旋偶合强度不同, 导致两种实验条件下加成产物上两个氢原子的能级不同。 在PASADENA条件即强磁场中, 氢原子的化学位移差大于其偶合常数, 同时p-H2两个氢原子核的自旋方向不同, 因此底物分子中两个超极化氢原子可取α β 和β α 两种自旋态。 在ALTADENA条件即弱磁场中, 两个氢的偶合常数大于化学位移差, 自旋-自旋偶合较强, 氢原子间自旋相关, 因此仅取β α 一种自旋态。 这导致两种实验条件下的加成产物呈现不同的增强氢谱: PASADENA条件, α β 和β α 自旋态可分别向α α 和β β 态跃迁, 产生两条倒峰和两条正峰[图3(c)]; ALTADENA条件下, β α 向α α 和β β 态跃迁, 产生一条倒峰和一条正峰[图3(d)]。
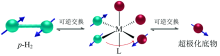
p-H2加成反应对底物有一定的要求, 即结构中存在如烯烃或炔烃等不饱和基团, 如无不饱和基团则需对底物进行化学修饰, 限制了此方法的底物范围。 另一类可行的方法是p-H2与底物在金属催化剂上通过可逆交换实现极化状态转移, 即可逆变换诱导信号增强(SABRE)[9, 10, 11]。 SABRE过程中p-H2和底物同时配位到极化转移催化剂, p-H2极化状态通过自旋-自旋偶合传至底物分子中核自旋量子数I=1/2的杂核原子[12]。 随后超极化底物分子从催化剂上脱离(图4), 可直接用于NMR谱学和成像[13]。 当然此类反应需要底物上存在能够配位到金属上的杂原子, 底物适用范围也存在一定的局限性。
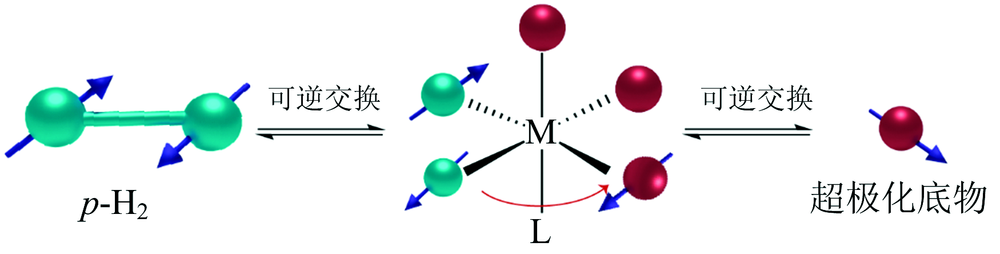 | 图4 通过SABRE将p-H2的超极化状态传递到底物分子的杂核原子Fig.4 Using SABRE to transfer the hyperpolarized spin-order of p-H2 to heteronucleus of substrates |
2.3节两种方法都可将超极化状态通过自旋-自旋偶合传递到异核原子, 传递极化效率由(JAX-JA'X)/(2JAA')决定, 其中ALTADENA和SABRE反应在低磁场下进行。 由于Jax-Ja'x小, 传递效率低, 因此可采用脉冲序列法和磁场循环法提高极化转移效率。
脉冲序列法通过持续提供氢原子核拉莫进动频率的脉冲照射, 减缓单重态的弛豫, 使底物分子的单重态不会存在较大损失[14, 15], 理论传递效率可接近100%, 脉冲序列结束后氢原子的超极化状态转到杂核原子[16, 17]。 此外, PASADENA[15, 18]或SABRE[19]实验也可通过脉冲序列将超极化状态转移到杂核原子上。 虽然PASADENA实验的理论传递效率仅为50%, 但此方法的优点是所有步骤整合在同一系统内, 操作简便[20, 21, 22, 23]。
第二种方法是磁场循环, 即加氢反应在地磁场中进行, 结束后迅速将超极化状态的底物分子转移到磁屏蔽容器。 底物分子感受到的磁场强度迅速降低, 原子核的拉莫尔进动频率大幅降低, 而核间J偶合依然存在。 由于异核振动频率与其偶合常数J相近, 经此绝热过程, 原子核的自旋能级发生各向同性混合并重排, 超极化自旋态转移到杂核原子[24]。 随后样品缓慢回到地磁场, 此过程为非绝热过程, 异核原子保持超极化状态[25]。 磁场循环法极化传递效率高, 但需额外仪器设备和操作步骤[26], 可用于远程偶合极化传递(J偶合常数≤ 1~2 Hz)[27]。
对PASADENA和ALTADENA, 仲氢加成反应效率对最终增强效果至关重要, 其中加氢催化剂的选择至关重要, 分为均相和异相加氢催化剂, 下面简单介绍此技术中常用的加氢催化剂。
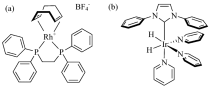
均相加氢催化剂通常采用铑金属配合物, 例如Rh(PPh3)3Cl和Rh(COD)(dppb)BF4等。 若采用多齿螯合磷配体, 例如双膦配体1, 4-双(二苯膦)丁烷(1, 4-bis(diphenylphosphino)butane, dppb), 能有效降低催化中间体的寿命[28], 提高H2加成反应速率, 因此Rh+(COD)(dppb)是目前最常用于p-H2加成反应的均相催化剂[图5(a)]。 此外, 将磷配体中的苯环换成丙磺酸基, 使催化剂可溶于水, 则底物范围拓展至水溶性底物分子, 除铑外还可采用基于钴的均相催化剂[29, 30]。 均相催化剂的生物毒性未知, 若用于生物成像需除掉催化剂。 采用分相法则无需除催化剂, 两相为有机相和水相, 加氢反应在有机相进行, 催化剂留在有机相, 不会对生命体产生毒性, 超极化底物进入水相[31, 32]。 更直接的方法是采用异相加氢催化剂, 不必考虑活体毒性问题, 只需将均相催化剂负载到固相[33, 34, 35], 或直接采用固体催化剂[36, 37, 38, 39, 40, 41]。
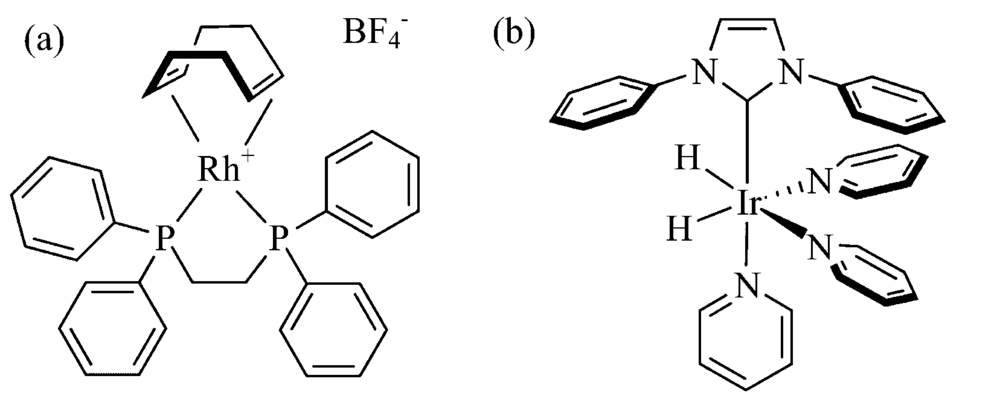 | 图5 (a)用于p-H2加成反应的均相催化剂, (b)用于SABRE的均相催化剂Fig.5 (a) Homogeneous catalyst for hydrogenation of p-H2, (b) homogeneous catalyst for SABRE |
SABRE实验中进行可逆交换的催化剂对最终超极化状态的传递效率至关重要, 首先H2可与金属中心配位, 其次配体交换反应的可逆性要好。 配位键为可逆非共价键, 其可逆性与金属中心、 周围配体及配体本身配位能力, 即配位键的强弱有关。 若配位键较弱, 则能配位到金属中心的底物比例低, 导致极化传递效率低。 若配位键过强, 则底物无法进行有效的可逆交换, 也会降低极化转移率。 对特定底物分子, 选择合适的可逆催化剂至关重要[42, 43], 目前常用于SABRE的催化剂是一类含铱的配合物[图5(b)], 其优点是稳定性高且底物适用范围广。 类似于加氢催化剂, 配体对可逆交换催化剂影响较大[44, 45], 可采用环辛二烯、 吡啶[46]、 卡宾[47, 48]等。 与异相加氢催化剂类似, 异相SABRE催化剂可通过将均相催化剂修饰到固相材料上制得, 固相材料可选高分子材料[49, 50]、 无机纳米颗粒[51]等; 也可通过分相法将催化剂留在有机相[52, 53, 54], 降低造影剂的生物毒性[55]。 可用于SABRE实验的催化剂除基于铱外, 还有基于钒[56]的异相催化剂及不含金属的催化剂体系[57, 58]。
常规NMR谱通常需要样品浓度大于mmol· L-1或采用较长的采集时间, 而PHIP和SABRE可将NMR灵敏度提高至少3个数量级, 可将所需浓度降为μ mol· L-1甚至更低, 这对NMR技术是质的飞跃, 为NMR应用与表征中间体结构、 痕量物质等低浓度体系开拓了新路。
在化学反应中, PHIP最早用于研究催化反应中间体, 催化中间体的结构对阐明催化路径、 解释催化机理并理性设计合成催化剂结构具重要意义。 尽管如此, 由于催化中间体含量少、 稳定性差及寿命短等特点, 其详细结构表征存在挑战。 借助仲氢诱导极化(PHIP), 超极化氢原子可作为NMR谱的“ 探针” , 结合NMR技术高分辨率的特点, 在短时间内能够给出丰富的分子结构和动力学信息, 因此非常适合研究催化反应过程。 Guan等采用此技术研究了一系列催化反应机理, 如图6所示[59], 发现Ir催化剂以H2作为还原剂, 将喹唑啉还原为3, 4-二氢喹唑啉, 通过增强NMR谱表征了Ir催化中间体的结构, 精确地确认了喹唑啉的还原位点, 并结合计算化学阐明此催化还原反应的路径和机理[60]。 Tokmic等借助PHIP研究Co催化剂将腈类化合物还原为伯胺的催化机理[61], 首先将氰基配位到Co(I)原子上, 氰基采用端基配位, 即氮的孤对电子与金属配位; 然后将H2加成到金属中心。 NMR监测表明p-H2成对加成到金属中心, 氰基的配位方式变为侧面配位, 即碳氮三键的π 轨道与Co配位, 这种配位方式使碳氮三键更容易被H2加成。
 | 图6 Ir催化剂将喹唑啉还原为3, 4-二氢喹唑啉的催化机理图Fig.6 Catalytic mechanism of hydrogenation of quinazoline into 3, 4-2H-quinazoline using Ir-based catalyst |
PHIP也可用于痕量物质检测。 Jeong等采用p-H2加成方法实现1 μ L苯乙烯在1 mL氯仿中的检测[62]。 在43 MHz核磁上仅需单次采集即可完成, 为今后NMR仪器的小型化提供可能。 Hermkens等利用SABRE技术实现水-乙醇混合物(例如威士忌)中类吡啶物质μ mol· L-1浓度的检测。 此技术操作简便, 与自制的试剂混合后可直接测试, 无需对待测样品进行提纯、 分离等操作[63]。 Hermkens等通过SABRE方法实现咖啡的甲醇提取液中吡啶和吡嗪的检测[64], 并借助SABRE将采集扩散排序(DOSY)谱所需的浓度降低至μ mol· L-1 [65]。
目前造影剂是生物核磁共振成像(magnetic resonance imaging, MRI)主要技术手段之一。 将造影剂注射/吸入活体内, 经由血管/气管到达成像部位, 通过MRI可检测造影剂的空间分布[66, 67, 68]。 目前文献中已报道多种基于p-H2超极化的造影剂, 其可分为以下几类: (1)惰性造影剂, 在活体中除基本物理行为(流动、 扩散)外, 不与活体发生特定的相互作用。 (2)新陈代谢造影剂[69, 70, 71], 超极化造影剂参与到活体新陈代谢。 (3)生理造影剂[72, 73], 对生理环境敏感, 可用于监测活体内pH值、 氧含量等关键生理参数。 (4)靶向造影剂[74], 用于研究特定的病理。
2.2.1 超极化造影剂
尽管PHIP能够显著提高造影剂在体外NMR信号, 但对活体内MRI信号的提升很有限, 主要是由于以下两点原因: (1)血液/组织液中存在大量水稀释造影剂, 同时这些水会产生很强的背景信号, 严重干扰造影剂成像信号, 导致活体内信噪比低。 (2)超极化造影剂极化寿命较短, 其注射/吸入到活体后需要一定时间才能到达特定成像区域, 在此过程中会因弛豫作用使信号变弱。 此外活体中存在的微量顺磁性物质如氧也会加快超极化造影剂的弛豫, 缩短极化寿命[75]。
为获得高质量活体图像, 超极化造影剂需满足下列条件: (1)活体毒性小, 能在活体内大量存在且不影响正常生理功能。 (2)弛豫时间长, 在到达特定成像区域后依然保持超极化状态。 (3)化学性质稳定, 除特定靶向物质外不与体内其他物质发生反应。 (4)对于液相生物医学MRI, 超极化造影剂应当具有较好水溶性, 以保证能够达到较高浓度。 使用超极化造影剂进行MRI检测可结合一些超快速成像技术, 例如自旋回波成像(echo planar imaging, EPI)、 时空编码(spatially encoded) MRI等技术, 在造影剂超极化状态消失前完成采样能提高MRI图像的信噪比。
2.2.2 液相磁共振生物医学成像
活体内存在大量液体环境(血液、 组织液和脑积液等), 许多重要生物化学反应在水中发生, 因此液相MRI是一种极其重要的生物医学成像方法, 制备相应的水溶性超极化造影剂就显得非常重要, 简单介绍三类液相造影剂。
2.2.2.1 新陈代谢造影剂
从柠檬酸循环中产生的琥珀酸盐(SUC)和还原型辅酶Ⅰ (NADH)的氧化过程驱动质子穿过线粒体内膜和ATP合成酶, 研究表明该途径的变化发生在几种疾病状态中。 13C-SUC可从前驱体二钠1-13C乙炔二羧酸酯(ADC)通过PHIP方法超极化得到, 并可通过氘代标记(富马酸-[1-13C]-2, 3-d2)延长弛豫时间(T1从6 s延长到27 s)。 为增加超极化造影剂的疏水性, 合成了衍生化二乙基-[1-13C]-富马酸-
Zacharias等成功制备超极化13C标记的琥珀酸(succinate, SUC)和琥珀酸二乙酯(diethyl succinate, DES), 均具有高极化率、 长T1弛豫时间、 低毒性和良好的水溶性等优点, 并在五种癌同种异体移植动物模型中研究了DES和SUC代谢过程, 包括乳腺癌(4T1), 肾细胞癌(RENCA), 结肠癌(CT26), 淋巴瘤NSO和淋巴瘤A20[71]。
 | 图7 携带RENCA(a)和淋巴瘤A20肿瘤的大鼠(b)的1H-MRI图像, 以及注射13C-HP DES(上色)后获得的13C MRI肿瘤的位置(黄色框表示)Fig.7 1H-MRI images of mouse with RENCA (a) and lymphoma A20 (b), and the location of tumor (yellow frame) after injected with 13C-HP DES (colored) |
2.2.2.2 生理造影剂
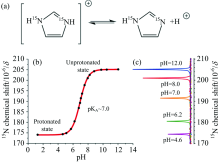
pH值失调是某些疾病的标志之一, 如过敏性哮喘、 动脉粥样硬化和肿瘤等, 研发pH响应的造影剂可实现基于不同pH值的成像。 咪唑-15N2可在80%水缓冲液中进行超极化[76], 咪唑-15N2的pKa值为7.0左右, 较大化学位移范围(约30 ppm)和低毒性使其成为体内pH成像的候选分子, 如图8(a, b, c)所示。 用于pH传感的杂核原子不限于15N, 也可拓展至其他异核。 如3-氟化吡啶对pH敏感, 且19F的信号范围大于10 ppm[77]。 由于19F具有较高的天然丰度、 旋磁比较高、 生物系统中可忽略不计的背景信号等优势, 因此基于19F的造影剂具重要研究价值。
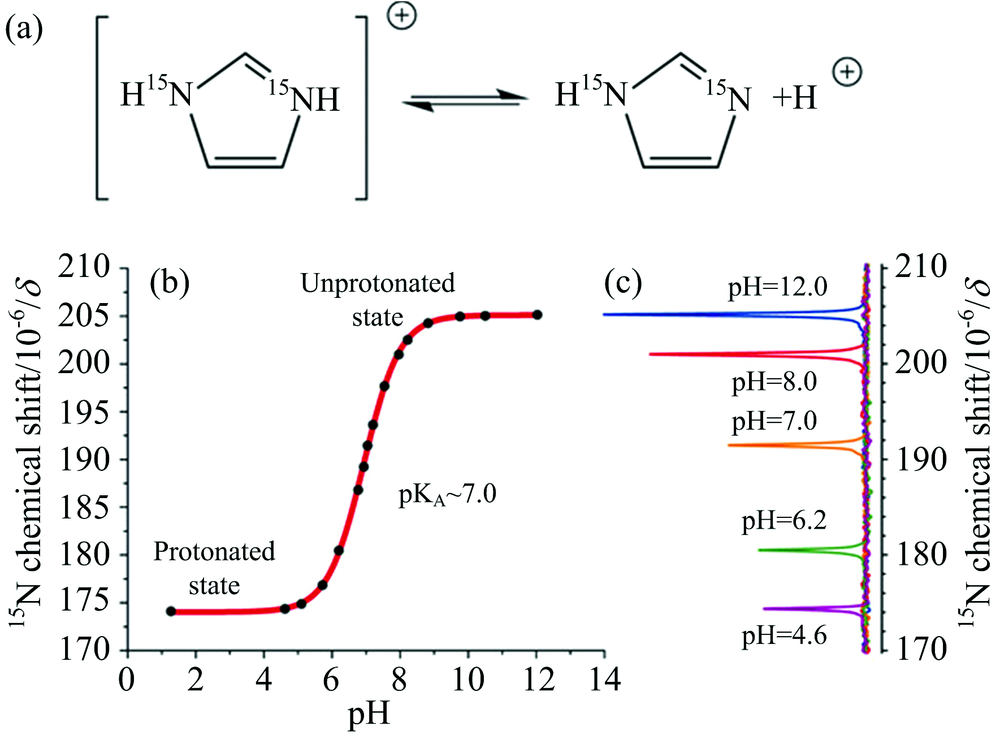 | 图8 (a)15N2-咪唑质子化的分子结构, (b)15N2-咪唑的化学位移与水溶液pH值间的关系, (c)不同pH值溶液中15N的化学位移[4]Fig.8 (a) Chemical structure of protonated 15N2-imidazole, (b) relationship between the chemical shift of 15N2-imidazole and the pH value of water, (c) the chemical shift of 15N in solution with different pH[4] |
缺氧是肿瘤分化、 侵入和放射治疗失败的一个重要预兆因素。 硝基咪唑的氧敏化合物常被用作缺氧造影剂, 当它进入到缺氧细胞时会与细胞内分子形成共价键而留在缺氧细胞内[73]。 15N-甲硝唑能够通过SABRE超极化过程生成高极化水平(大于34%)的底物分子[54]。
2.2.2.3 靶向造影剂
超极化靶向分子造影剂在MRI领域报道较少, 但由于其在生命体内的高选择性而极具研究价值。 首例基于受体成像(如正电子发射断层扫描)的PHIP靶向分子探针是丙酸四氟丙酯-1-13C-d3(TFPP)[74]。 TFPP具有丙酸盐类似的化学结构, 可与脂质双分子层结合后纵向弛豫时间减少[T1=(21± 1) s]。 与正常喂养的对照组相比, 在动脉稠化的小鼠体内观察到13C-HP TFPP在完整心脏心内膜表面上结合紧密, 表明TFPP可用于动脉稠样硬化斑块的体内成像分子探针, 可作为具有增强MR敏感的受体靶向生物成像模型。
2.2.3 气相磁共振生物医学成像
活体中存在大量气体环境(肺部、 气管等), 对含气体较多组织的MRI需要特定的气体超极化造影剂。 目前正着重研究开发除昂贵惰性气体外的气体超极化技术, 例如3He和129Xe等, 如Ruan等采用DNP超极化的氙气作为肺部造影剂[78]。 PHIP超极化气态烃是有前途的替代方案之一[79], 例如超极化丙烷由丙烯与p-H2加成制备, 并成功应用于模型物体成像以及原位微型反应器成像。 Kovtunov等将一系列金属负载到TiO2基底上作为加氢催化剂, 实现超极化丙烷的三维核MRI[80]。 由丙炔生成超极化丙烯需要采用选择性催化剂, 例如Pd-In/Al2O3对丙烯的选择性高达98%, 能显著增强极化产物信号(对丙烯的CH基团信号增强3 400倍, 相当于极化率达到9.4%), 丙炔的转化率高达18%, 能够获得超极化丙烯的1H-MRI图像, 该方法还可以应用于选择性催化氢化反应的原位可视化MRI技术中。
仲氢诱导增强超极化技术是一种低成本、 高效率实现超极化的NMR方法, 原料p-H2可通过将H2降温后加催化剂制得, 通过两种化学反应可简便高效地得到超极化底物分子, 已经在化学和生物成像等领域得到初步应用, 目前的研究重点在多种催化剂和配体的选择, 以及仪器设备自动化方面。 虽然仲氢诱导增强极化相关研究还处于早期阶段, 但其已经展现巨大的应用潜力, 在不远的将来一定会在化学和生命研究等学科发挥越来越重要的作用。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|
| [80] |
|