{kind=link}
{kind=link}
{kind=link}
{kind=link}
氨基酸官能团的太赫兹振动模式研究
[燕芳 , 李伟
, 李伟* , 王志春]
, 李伟, 王志春]
|
|


作者简介: 燕 芳, 1980年生, 内蒙古科技大学信息工程学院副教授 e-mail: 0472yanfang@163.com
对比于氨基酸的红外分析法, 太赫兹波的电子能量更低, 可实现无损检测。 氨基酸分子内原子振动、 分子间氢键的作用、 以及晶体中晶格的低频振动均处于太赫兹波段, 使其在太赫兹波段具有吸收峰, 且不同的氨基酸分子太赫兹吸收峰不同, 故可用氨基酸在太赫兹波段的这种“指纹特性”实现氨基酸类物质的定性分析。 量子化学分析方法可以应用量子力学的基本原理和方法, 研究稳定和不稳定分子的结构、 性能及其之间的关系, 还可以针对分子与分子间的相互作用、 相互碰撞及相互反应等问题进行研究。 通过量子化学计算方法计算氨基酸分子的太赫兹吸收谱, 可以为氨基酸分子的太赫兹吸收峰匹配分子振动模式, 对氨基酸定性分析有一定参考性与指向性, 并为实验获取的样品太赫兹时域光谱提供理论支撑, 在实验获得太赫兹吸收谱的基础上进行量子化学计算, 还能为实验结果进行验证。 首先利用太赫兹时域光谱技术获取了谷氨酰胺、 苏氨酸、 组氨酸的太赫兹吸收谱, 分别构建这三种氨基酸样品在实物中以两性离子形式存在的单分子构型, 利用量子化学计算方法在完成结构优化后进行太赫兹吸收谱模拟计算。 计算结果表明三种氨基酸单分子的太赫兹吸收谱计算结果与实验获取的太赫兹吸收谱差异较大, 但在高频段吸收峰峰位基本吻合。 通过GaussView分别查看了这三种氨基酸分子在太赫兹段内的吸收峰对应频率处的振转情况, 发现在高频段内三种氨基酸分子官能团均只发生转动而未见振动, 并且转动模式基本一致。 通过对氨基酸官能团的太赫兹吸收谱进行量子化学计算, 将官能团在高频段内吸收峰对应频率处的振转模式与三种氨基酸分子在该段内吸收峰对应频率处的振转模式做了对比。 研究表明, 在氨基酸单分子构型下由量子化学方法计算所得的太赫兹吸收谱中, 高频段内计算得出的模拟吸收峰与实验获取的太赫兹吸收峰基本吻合; 振转模式分析发现, 谷氨酰胺、 苏氨酸、 组氨酸在太赫兹高频段内的氨基酸官能团振转模式相同, 三种氨基酸分子在高频段内的吸收峰主要来源于氨基酸官能团。 因此, 结合量子化学计算与太赫兹吸收谱可以实现氨基酸类物质的定性分析。
Compared with the infrared analysis of amino acids, terahertz wave has lower electronic energy and can be used for nondestructive testing. The intramolecular atomic vibration, the intermolecular hydrogen bond and the low-frequency vibration of the crystal lattice of amino acids are all in terahertz band, which makes them have absorption peaks in terahertz band, and different amino acid molecules have different Terahertz Absorption peaks. Therefore, this fingerprint characteristic of amino acids in terahertz band can be used for qualitative analysis of amino acids. Quantum chemical analysis methods can apply the basic principles and methods of quantum mechanics to study the structure, properties and relationships of stable and unstable molecules. It can also study the interactions, collisions and reactions between molecules. The Terahertz Absorption Spectra of amino acids can be calculated by quantum chemical calculation method, which can match the molecular vibration mode of the terahertz absorption peaks of amino acids. It has certain reference and directivity for the qualitative analysis of amino acids, and provides theoretical support for the terahertz time domain spectra of samples obtained from experiments. Quantum chemical calculation is carried out on the basis of the terahertz absorption spectra obtained from experiments. It can also validate the experimental results. In this paper, Terahertz Absorption Spectra of glutamine, threonine and histidine were obtained by terahertz time-domain spectroscopy. The monomolecular configurations of these three amino acids in the form of amphoteric ions were constructed respectively. The Terahertz Absorption spectra were simulated by quantum chemical calculation method after the structural optimization was completed. The calculated results showed that the Terahertz Absorption Spectra of three amino acids are quite different from those obtained by experiments, but the peak positions at high frequencies are basically the same. GaussView was used to observe the vibration and rotation of the absorption peaks of the three amino acids at the corresponding frequencies in the terahertz band. It was found that the functional groups of the three amino acids only rotated without vibration in the high frequency band, and the rotation modes were basically the same. The Terahertz Absorption Spectra of amino acid functional groups were calculated quantum chemistry. The vibration and rotation patterns of functional groups at the corresponding frequencies of the absorption peaks in the high frequency band were compared with those of three amino acid molecules at the corresponding frequencies of the absorption peaks in the high frequency band. The results showed that in the terahertz absorption spectra calculated by quantum chemistry method under the single molecular configuration of amino acids, the simulated absorption peaks calculated in the high frequency band are basically consistent with the experimental Terahertz Absorption peaks. Vibration mode analysis showed that the vibrational modes of amino acid functional groups of glutamine, threonine and histidine were the same in the terahertz high frequency band. The absorption peaks of the three amino acid molecules in the high frequency band mainly came from amino acid functional groups. Therefore, the qualitative analysis of amino acids can be realized by combining quantum chemical calculation with Terahertz Absorption spectrum.
太赫兹波的能量介于光子和电子之间, 能穿透非金属和非极性材料, 使太赫兹时域光谱技术(THz-TDS)作为无损检测的一项新兴技术手段成为可能。 许多生物分子的集体振转模式位于太赫兹波段, 使生物分子在太赫兹波段具有指纹性, 获得待测物质的太赫兹吸收谱后, 与标准谱进行对照可以实现对待测物质的定性分析。 在此基础上, 结合最小二乘法、 支持向量机等数据处理技术还可以实现基于太赫兹时域光谱对待测物质的定量分析。
构成蛋白质的二十种氨基酸与生命活动紧密相关, 王卫宁等[1]研究发现, 氨基酸类物质在实验的有效光谱范围内, 具有各自的特征吸收, 因此可以利用太赫兹时域光谱技术定性鉴别物质, 并对精氨酸、 组氨酸的太赫兹吸收谱与分子间集体振转模式的对应关系做了相关研究。
目前的量子化学分析计算在国际上广泛使用Gaussian03软件。 Gaussian03基于从头算理论、 半经验法和密度泛函等计算方法, 在气相范围内研究分子结构及能量、 分子间作用力、 IR和拉曼光谱等具有突出优势[2]。 研究生物分子太赫兹吸收峰的归属, 多采用密度泛函理论, 为不同的分子匹配不同的算法、 不同的基组(如杂化泛函算法、 6-311G++基组)计算生物分子的红外振动谱, 为在红外谱内提取的太赫兹谱做简正振动分析, 对分子结构内振转与太赫兹吸收峰做出指认。
在对氨基酸样品的太赫兹吸收谱计算中, 为了分析与样品更为接近的构型, 应选择全面反映分子间作用力的晶胞构型作为计算的初始构型。 而晶胞构型计算对数据处理器的要求偏高, 同时运算时间较长(30 h以上), 限于以上缺点该方法对于大量样品的计算适用性不强。 本文通过对三种氨基酸样品单分子构型的分析, 找到了吸收峰所在频率对应的原子或原子团振转模式的一致规律, 可以为氨基酸类物质的太赫兹定性分析提供一定的参考。
利用透射式太赫兹时域光谱系统获取谷氨酰胺、 苏氨酸和组氨酸样品的太赫兹吸收谱, 设备详细描述见文献。 通过在密闭的实验光路中充满氮气来降低环境湿度对太赫兹波的吸收[3], 测试温度为室温25 ℃。 吸收谱的获取参照了Dorney等[4]提出的方法。
为了降低颗粒度差异引起的太赫兹波散射, 实验所用谷氨酰胺、 苏氨酸、 组氨酸样品经充分研磨后加入120 mg聚乙烯粉末混合, 以8 MPa压力冲压成片, 样片厚度约0.9 mm, 直径13 mm。
实验中, 分别记录太赫兹光路中有无样品时的时域信号作为样品信号和参考信号, 为了减小 THz-TDS系统测量时域样品信号和参考信号的误差, 每种样品均测量三次后取其平均值。 然后对时域测量结果做傅里叶变换, 样品信号傅里叶变换结果记为Esam(ω ), 参考信号傅里叶变换结果记为Eref(ω )。 取Esam(ω )和Eref(ω )比值的幅频特性和相频特性, 如式(1)和式(2)所示
其中: d为样品厚度, c为光速, ω 为角频率, ϕ (ω )为相位差。 折射率n(ω )和吸收系数α (ω )为
由式(4)即可计算出样品的太赫兹吸收谱。
量子化学分析计算在Gaussian03软件中进行, 采用密度泛函理论中的杂化泛函理论, 为不同分子选用了不同基组, 首先, 对谷氨酰胺、 苏氨酸、 组氨酸样品进行单分子构型计算, 为了对氨基酸类物质进行定性分析, 还构建了氨基酸官能团的模型并进行了相应的量子化学计算。
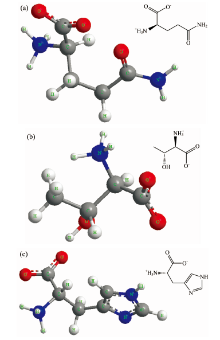
氨基酸在固体样品中以两性离子形式存在, 故将对谷氨酰胺、 苏氨酸、 组氨酸单分子的两性离子进行计算分析。 分子构型在ChemDraw中绘制, 导出分子结构的笛卡尔坐标并导入Gaussian03进行运算。 因两性离子带正负两性电荷, 故选用6-311G++(d, p)基组, 同时采用基于B3LYP杂化泛函的密度泛函理论做构型优化[5], 最后进行IR计算。 由于太赫兹波处于远红外波段, 故太赫兹吸收谱需在IR的远红外段提取。 图1(a), (b)和(c)分别为谷氨酰胺、 苏氨酸、 组氨酸两性离子形式存在的单分子构型。
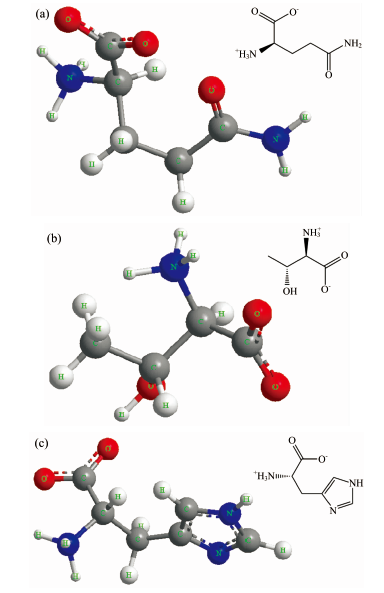 | 图1 三种氨基酸样品的单分子构型 (a): 谷氨酰胺单分子构型; (b) 苏氨酸单分子构型; (c): 组氨酸单分子构型Fig.1 Single molecular configuration of three amino acid samples (a): Single molecular configuration of glutamine; (b): Single molecular configuration of threonine; (c): Single molecular configuration of histidine |
三种样品的单分子构型在量子化学计算过程中, 选用的运算理论与基组均相同, 计算机时与收敛迭代次数限于原子个数、 分子空间结构和数据处理器配置。 三种样品的计算机时和收敛迭代次数示于表1, 计算所用数据处理器配置为2.6 GHz主频, 单核四线程, 缓存数据存储器为128 G固态硬盘(SSD)。
| 表1 计算机时及收敛迭代次数 Table 1 Computing time and convergence iteration number |
王卫宁等指出, 发展较为成熟的红外吸收谱的构成是由固定的原子或原子基团的峰位、 峰形支撑的。 也就是说, 各原子或原子基团有特定的红外吸收峰位和峰形。 保持氨基酸类物质化学通性的最小基团为氨基酸官能团, 为了探索太赫兹吸收谱是否具有和红外吸收谱相类似的性质, 研究氨基酸官能团在太赫兹波段是否具有固定峰位, 本文构建了氨基酸官能团的单分子模型, 计算了氨基酸官能团的太赫兹吸收谱。
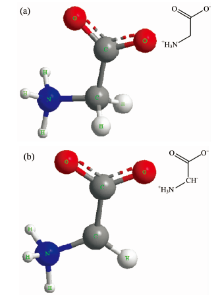
氨基酸官能团由一个氨基和一个羧基构成, 为了与氨基酸固体样品做出更精确的对照, 将氨基酸官能团也绘制成两性离子。 而且, 绘制了含R基和不含R基两种结构, 其中含R基结构用一个氢(H)原子取代R基, 不含R基为C-带电体形式。 计算过程与三种样品的单分子计算类似, 由ChemDraw绘制分子构型, 导出笛卡尔坐标后, 在Gaussian03中进行优化和IR计算。 其中氢原子取代R基的氨基酸官能团选用6-311G++(d, p)基组, 而不含R基的氨基酸官能团带两个单位负电荷和一个单位正电荷, 在基组选择时考虑到不含R基的氨基酸官能团电荷个数为3, 但依然带两性电荷, 故仍然选用6-311G++(d, p)基组。 图2(a)和(b)分别为含R基和不含R基氨基酸官能团的单分子构型。
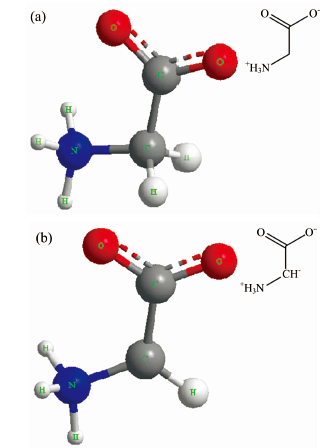 | 图2 氨基酸官能团的单分子构型 (a): 含R基氨基酸官能团; (b) 不含R基氨基酸官能团Fig.2 Single molecular configuration of amino acid functional groups (a): Functional groups containing R-based amino acids; (b): Functional groups without R-based amino acids |
利用量子化学分析软件计算IR后, 在远红外波段(波数3~333 cm-1)提取太赫兹吸收谱, 由于实验所用仪器精度所限, 3 THz后的实验数据受环境因素影响较大, 故只提取3 THz(波数3~100 cm-1)之前的计算数据与有效的实验数据进行对比。
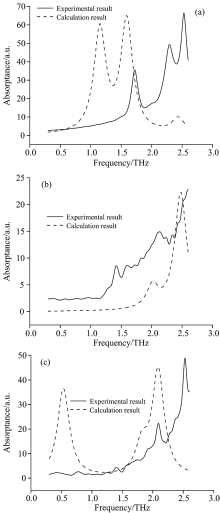
图3(a), (b)和(c)中分别对比了三种氨基酸分子的实验谱和由量子化学计算获得的模拟谱。
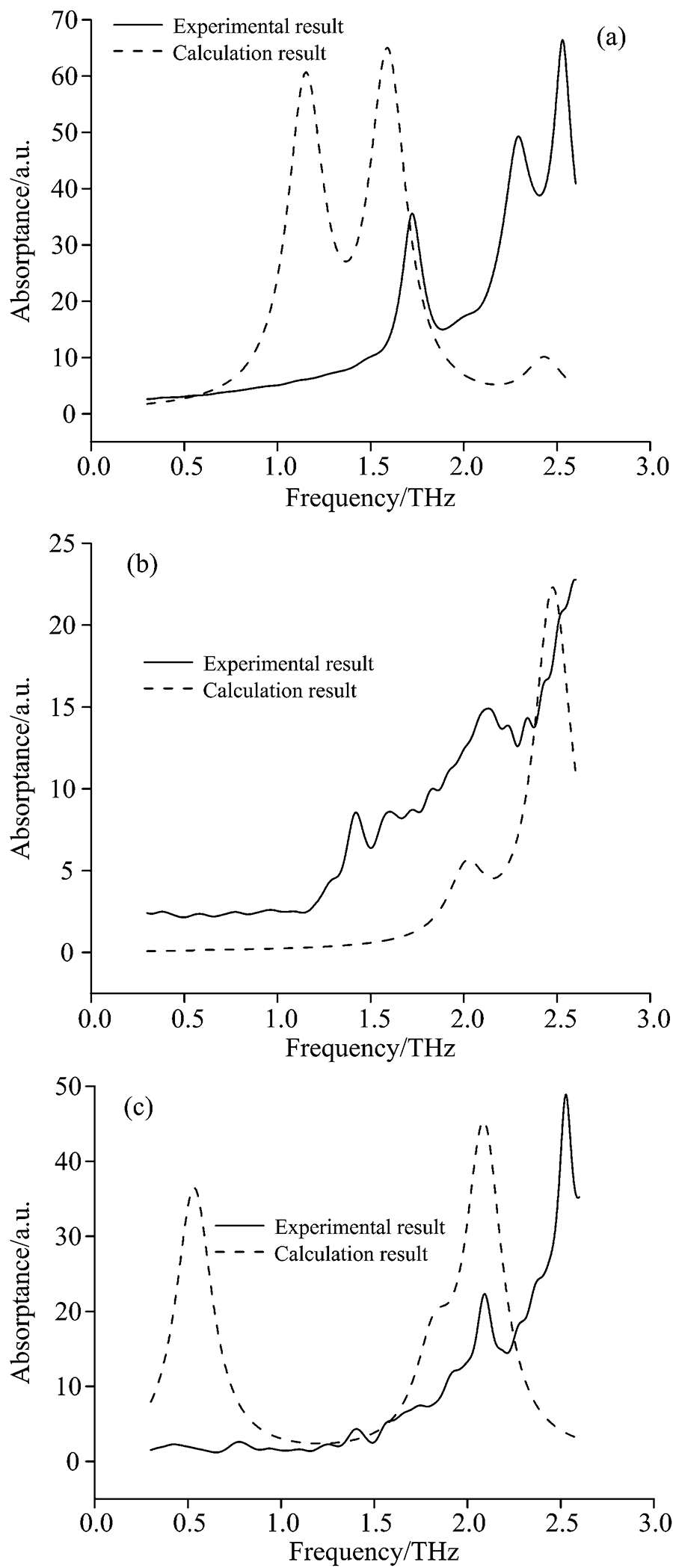 | 图3 三种氨基酸的实验和计算数据对比 (a): 谷氨酰胺实验谱与计算谱对比; (b): 苏氨酸实验谱与计算谱对比; (c): 组氨酸实验谱与计算谱对比Fig.3 Comparison of experimental and computational data for three amino acids (a): Comparison of experimental and computational spectra of glutamine; (b): Comparison of experimental and computational spectra of threonine; (c): Comparison of experimental and computational spectra of histidine |
对比发现, 每种氨基酸由量子化学计算获得的模拟谱在高频段内的吸收峰与实验谱基本一致, 而低频段吸收峰的峰位和峰形均与实验谱有较大差距。 这是由于氨基酸单分子构型的计算仅提供了分子内部原子及原子基团在太赫兹频段内的振转结果, 没有体现物质内分子间的氢键及范德华力作用, 而分子间氢键及范德华力的作用在太赫兹频段内的振转结果集中体现于实验获得的太赫兹吸收谱低频段内。
不含R基的氨基酸官能团整体电性为一个单位负电荷, 在体系中不稳定, 自然界中不存在, 量子化学优化结果不收敛(找不到体系能量最低点), 5 THz以下的太赫兹频段未见吸收峰, 5.3 THz时出现第一个吸收峰。 含R基的氨基酸官能团整体为电中性, 可以找到体系总能最低点, 2.8 THz时出现第一个太赫兹吸收峰, 但由氢原子取代R基的官能团只能保持氨基酸的部分理化性质。
研究发现三种氨基酸样品的实验谱与模拟谱在2~3 THz基本吻合, 含R基官能团在2.8 THz也出现吸收峰。 为了讨论氨基酸类物质2~3 THz的吸收峰是否由氨基酸官能团内原子的振转提供, 接下来为三种样品单分子构型和H-氨基酸官能团单分子构型在2~3 THz内吸收峰对应频率的振转模式做了指认。
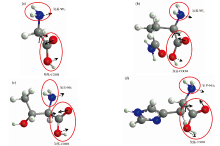
由于H-氨基酸官能团不能完全代表实际氨基酸官能团的理化性质, 对H-氨基酸官能团与谷氨酰胺、 苏氨酸、 组氨酸在2~3 THz内的振转模式的指认如图4所示。
 | 图4 氨基酸官能团振转模式指认 (a): H-氨基酸官能团2.8 THz振转图; (b): 谷氨酰胺内官能团2.41 THz振转图; (c): 苏氨酸内官能团2.49 THz振转图; (d): 组氨酸内官能团2.09 THz振转图Fig.4 Identification of oscillation and rotation patterns of amino acid functional groups (a): 2.8 THz oscillation diagram of the internal functional groups of H-amino acid; (b): 2.41 THz oscillation diagram of the internal functional group of glutamine; (c): 2.49 THz oscillation diagram of the internal functional group of threonine; (d): 2.09 THz oscillation diagram of the internal functional group of histidine |
指认结果显示, 在该频段内氨基酸官能团不发生振动, 只存在C— C键旋转, 旋转角约为60° 。 C— C键旋转使氨基由图示位置顺时针转动, 羧基由图示位置逆时针转动。
谷氨酰胺、 苏氨酸、 组氨酸单分子构型的太赫兹吸收谱计算结果在2~3 THz段内的吸收峰与实验吸收谱在该频段的结果基本一致, 但其他吸收峰差距较大, 表明样品的太赫兹吸收谱是分子内原子及原子团作用、 分子间氢键及范德华力作用的集中体现。 另外, 含R基氨基酸官能团在2~3 THz段的吸收峰为2.8 THz, 而且在该段内, 含R基官能团振动模式与谷氨酰胺、 苏氨酸、 组氨酸官能团振转模式只发现C— C键的旋转。 氨基酸官能团在2~3 THz段内的振转是氨基酸类物质在该段内吸收峰的主要来源, 据此可以实现氨基酸类物质的定性指认。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|