
{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于密度泛函理论的对二甲苯分子电子光谱的外场效应研究
[张倩1  , 杜建宾
, 杜建宾1, 2, * , 冯志芳3 , 张亚如1 , 唐延林4 ]
, 杜建宾, 冯志芳|
|


作者简介: 张倩, 女, 1985年生, 廊坊师范学院物理与电子信息学院讲师 e-mail: 514520923@qq.com
对二甲苯(PX)是化工领域一种非常重要的原料, 被广泛地用于香料、 医药、 油墨和农药等的生产, 因此研究PX分子的电子光谱和外场效应, 对于它的检测和降解具有十分重要的意义。 为研究外电场作用下, PX分子的紫外-可见(UV-Vis)光谱的变化, 采用密度泛函理论(density functional theory, DFT)B3LYP方法在6-311++G(d, p)基组水平上, 优化了不同外电场(0~0.025 a.u., 0~1.285 6×1010 V·m-1)作用下PX分子的基态几何构型, 在此基础上利用含时密度泛函理论(TDDFT)计算了PX分子的UV-Vis吸收光谱, 最后对PX分子紫外吸收峰和摩尔吸收系数受外电场作用的的影响规律进行了研究。 结果表明: 有波长为189 nm、 摩尔吸收系数为35 580 L·mol-1·cm-1的强吸收峰, 处于 E1带, 它是环状共轭的三个乙烯键的苯型体系中的π→π*电子跃迁产生的; 与苯分子相比, 吸收峰出现11 nm的红移: 由于两个甲基和苯环形成p-π共轭, 苯环的大π键变弱, 故PX分子的紫外吸收峰出现红移; 当增加了外电场后, 最低未占据轨道(LUMO)向外电场的反方向偏移, 导致苯环上的电子密度减小, 大π键变弱, π→π*跃迁需要的能量降低, 电子跃迁产生的波长增大, 吸收峰出现显著红移, 当外电场增大到0.020 a.u.时, 红移已经非常明显; 外电场的引入, 导致苯环上的电子密度减小, 大π键变弱, π→π*跃迁的电子数减少, 摩尔吸收系数降低, 随着外电场的增强, 摩尔吸收系数降低明显, 尤其在外电场增强到0.020a.u.后, 摩尔吸收系数降低非常显著。 这些工作为PX的检测和降解方法研究提供了一定的理论依据, 也对其他有机污染物的检测方法和降解机理的研究有启示作用。
Para-xylene(PX) is an important raw material in chemical industry. In order to study the change of ultraviolet-visible(UV-Vis) spectra of PX under external electrical field, the density functional theory (DFT) has been employed to calculate geometrical parameters of the ground state of PX molecule under different external electric fields ( from 0 to 0.025 a.u.) in this paper. On this basis, the UV-Vis absorption spectra of PX molecule were calculated by employing the time-dependent density functional theory (TDDFT). At last, by employing the TDDFT in the same fundamental group, we studied wavelength and the molar absorption coefficient of the first twenty-six excited states of PX molecule under different external electric fields. The results show that the most absorption of UV-Vis absorption spectra appears in the E1 belt of benzene electronic transition from π to π*; the ultraviolet absorption peaks of excited states of PX are proved to appear observably red shift and the molar absorption coefficient sharply decreases with the increase of the field intensity.
对二甲苯(para-xylene, PX)作为聚酯行业的重要原料, 主要用于制造精对二苯甲酸或精对二苯甲酸二甲酯, 进而生产聚酯[1], 还可用作溶剂以及合成纤维、 材料等行业[2]。 长期接触对二甲苯, 会导致肝、 肾损伤, 严重时会危及生命。 张冰洁等研究了PX对人类肝细胞的毒性效应[3], 刘洋等对微孔活性炭对PX的吸附和脱附性能进行了研究[4], 林华等探讨了外电场对PX结构和光谱特性的影响[5], 但对于PX在外电场下的电子光谱的深入研究目前还未见报道。 本文采用含时密度泛函理论(time-dependent density functional theory, TDDFT)[6, 7, 8]对不同外电场下PX分子的紫外-可见(ultraviolet-visible, UV-Vis)光谱的变化进行了研究, 这为以PX为原料合成的有机污染物的降解和检测方法研究提供了理论依据。
分子体系的哈密顿量H当有外电场时加入由两项组成[9], 即无电场时的哈密顿量H0和外电场与分子体系相互作用的哈密顿量Hint
Hint在偶极近似下可以表示为
其中μ 为分子的电偶极矩。
PX分子的结构如图1所示, 在沿x轴方向上加上一系列强度为0~0.025 a.u.(0~1.285 6× 1010 V· m-1)的外电场, 即在Gaussian程序的哈密顿量中添加$H_{int}$[10-11], 然后利用TDDFT来精确计算分子前26个激发态的波长和摩尔吸收系数在外电场作用下产生的变化。 全部计算在Gaussian09软件包中完成。
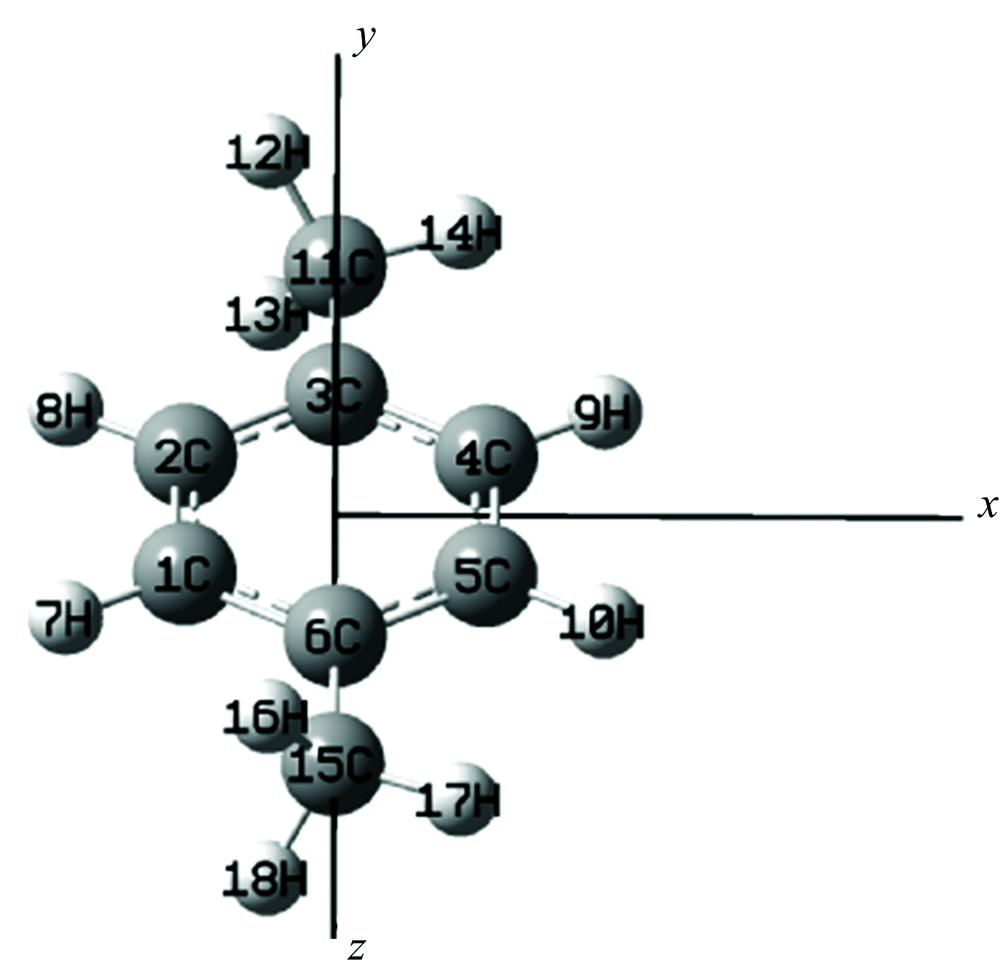 | 图1 PX分子结构示意图Fig.1 Optimized molecular structure of PX |
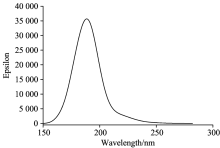
在分子基态几何结构优化的基础上, 采用含时密度泛函理论(time-dependent density functional theory, TDDFT)方法计算了PX分子的UV-Vis吸收光谱, 如图2所示, 有一个波长为189 nm、 摩尔吸收系数为35 580 L· mol-1· cm-1的强吸收峰, 处于E1带, 它是环状共轭的三个乙烯键的苯型体系中的π → π * 电子跃迁产生的。 在NIST Chemistry WebBook(国际标准与技术学会化学电子数据库)中给出了苯分子165~205 nm波长范围的UV-Vis吸收光谱, 其最强吸收峰出现在波长178 nm处。 PX是苯分子对位的两个H被两个甲基取代所形成, 这两个给电子基团和苯环形成p-π 共轭, 苯环的大π 键变弱, 分子的紫外吸收峰出现红移, 与理论的计算结果是一致, 说明理论计算的PX分子UV-Vis吸收光谱是可信的。
 | 图2 无外电场时PX分子的UV-Vis吸收光谱Fig.2 UV-Vis absorption spectra of PX without external electric fields |
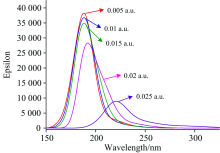
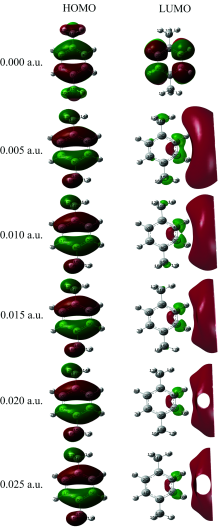
在PX分子基态优化的基础上, 采用TDDFT方法研究了0~0.025 a.u.的外电场对分子激发态波长λ 和摩尔吸收系数的产生影响, 如图3所示, 可以看出, 当外电场较弱时, 吸收峰的波长变化不明显, 但随着外电场的增强, 吸收峰出现显著红移, 当外电场增大到0.020 a.u.时, 红移已经非常明显, 这是由于无外电场时, 分子中的电子轨道主要分布在苯环上(如图4所示), 当增加了外电场后, LUMO轨道向外电场的反方向偏移, 导致苯环上的电子密度减小, 大π 键变弱, π → π * 跃迁需要的能量降低, 电子跃迁产生的波长增大, 因此出现吸收峰红移。 当外电场增大到0.020 a.u.时, LUMO偏移更加明显, 出现了电子密度为0的区域, 因此吸收峰也出现了显著红移。
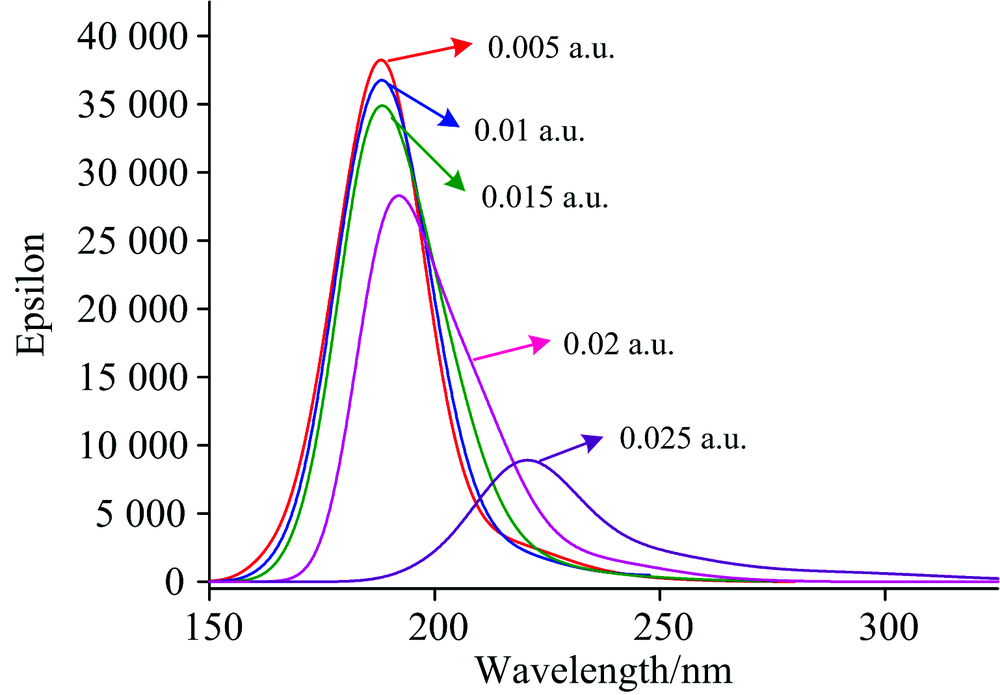 | 图3 PX分子在不同外电场作用下的UV-Vis吸收光谱Fig.3 UV-Vis absorption spectra of PX under different external electric fields |
 | 图4 不同外电场下PX激发态的分子前线轨道 HOMO: 最高占据轨道; LUMO: 最低未占据轨道Fig.4 Excited state frontier orbital diagram of PX molecular under different external electric fields HOMO: Highest occupied molecular orbital; LUMO: Lowest unoccupied molecular orbital |
随着外电场的增强, 吸收峰的红移, 摩尔吸收系数降低明显, 尤其在外电场增强到0.020 a.u.后, 摩尔吸收系数降低已经非常显著, 这是由于外电场作用使电子云的整体偏移, 使得LUMO偏离苯环, 苯环上的电子云密度变小(如图4所示), π → π * 跃迁的电子个数减少, 从而摩尔吸收系数降低。
基于密度泛函理论研究了不同外电场对PX分子的激发态波长和摩尔吸收系数的影响。 结果表明, 无外电场时, 分子最强吸收峰的波长为189 nm, 由环状共轭的三个乙烯键的苯型体系中的π → π * 电子跃迁所产生; 随着外电场的增强, 分子的紫外吸收峰明显红移, 摩尔吸收系数显著下降。 综上分析可见, 在外电场的作用下, 分子的UV-Vis吸收光谱变化显著。 与塑化剂类分子的外场效应对比, 苯环上连有长支链的有机物对外电场的变化更加敏感[7]。 这些工作对PX的检测和降解方法研究提供了一定的理论依据, 也对其他有机污染物的检测方法和降解机理的研究有启示作用。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|