{kind=link}
{kind=link}
{kind=link}
荧光光谱法研究氨基改性介孔泡沫对DhaA的稳定化机理
[郑禾 , 钟近艺
, 钟近艺* , 崔燕, 赵冲林, 郭旋, 吴琼, 赵渊中, 娄雷]
, 钟近艺, 崔燕, 赵冲林, 郭旋, 吴琼, 赵渊中, 娄雷]
|
|


作者简介: 郑禾, 1984年生, 军事科学院防化研究院助理研究员 e-mail: fhyjyzh@126.com
采用F-4600荧光光谱仪, 对尿素和二甲基亚砜两种变性剂中烷基卤脱卤酶DhaA在氨基改性介孔泡沫固定化前后的荧光光谱特征进行测定。 运用荧光相图分析DhaA在两种变性剂中的去折叠过程, 并结合活性残留率进行了变性过程热力学参数计算, 比较固定化前后DhaA去折叠过程和热力学参数的区别。 实验结果表明, DhaA催化活性随变性剂浓度增加而降低。 相同变性剂浓度下, 固定化DhaA能够比游离态DhaA保持更高的催化活性, 在变性剂到达临界浓度之前(尿素浓度5.5 mol·L-1, DMSO浓度7 mol·L-1), 氨基改性介孔泡沫的稳定化作用显著。 DhaA在尿素诱导下的变性过程符合“二态模型”, 而在DMSO诱导下符合“三态模型”, DhaA中间态出现在浓度为5.6 mol·L-1。 氨基改性介孔泡沫固定化不改变DhaA变性过程, 但能够提高DhaA的去折叠热力学参数。 在尿素诱导下, 计算得到的DhaA初始吉布斯自由能变Δ G(H2O)为8.51 kcal·mol-1, 固定化后Δ G(H2O)提高为9.55 kcal·mol-1; 但由于尿素分子容易通过静电作用进入氨基介孔泡沫孔道, 固定化后DhaA的溶液可及面积 m由3.69 kcal·(mol·mol·L-1)-1增大到4.00 kcal·(mol·mol·L-1)-1, 孔道内的氨基、 羟基能够通过氢键作用增强DhaA的刚性, 从而有效的降低了尿素可及面积增加带来的影响, 提高了DhaA的尿素耐受性。 在DMSO诱导下, 计算发现游离态与固定化DhaA在折叠态向中间态转变过程中的Δ G(H2O)均为12.12 kcal·mol-1, 由于孔道内的氨基、 羟基能够有效阻碍非极性DMSO分子的进入, 造成m从3.39 kcal·(mol·mol·L-1)-1降低为2.30 kcal·(mol·mol·L-1)-1; 当DhaA从中间态向去折叠态转化时, DhaA内部疏水基团暴露导致 m增加, 由于孔道内极性微环境作用, 固定化DhaA的 m值(4.40 kcal·(mol·mol·L-1)-1)仍然低于游离态DhaA(4.94 kcal·(mol·mol·L-1)-1)。 荧光光谱法研究固定化对DhaA去折叠过程及热力学参数的影响是深入研究DhaA稳定性的有效手段, 能够为其他生物酶的稳定化机理研究提供方法指导。
The fluorescence spectroscopic characteristics of DhaA immobilized by amino-modified-mesocellular foam (MCF-NH2) were investigated during denaturation procedures with urea and DMSO as denaturant by using F-4600 fluorescence spectrometer. The unfolding process of DhaA in denaturants was analyzed by phase diagram of fluorescence, and thermodynamic parameters were calculated with residual activity ratio of DhaA. The difference between the non-immobilized and immobilized DhaA in aspect of unfolding process and thermodynamic parameters were compared. The results showed that the catalytic activity of DhaA declined with the increasing concentration of denaturants, and the catalytic activity of immobilized DhaA could be maintained better than that of free DhaA at the same concentration of denaturant. The stabilization effect of MCF-NH2 to DhaA was obvious before the denaturants concentration reached a critical concentration (5.5 mol·L-1 for urea, 7 mol·L-1 for DMSO). The unfolding procedure of DhaA induced by urea conformed to typical “two-state” model. The unfolding procedure of DhaA induced by DMSO conformed to “three-state” model, and the intermediate state of DhaA appeared at the DMSO concentration of 5.6 mol·L-1. Immobilization by MCF-NH2 didn’t change the degeneration process of DhaA, but could raise the thermodynamic parameters during the unfolding process of DhaA. When induced by urea, the Δ G(H2O) was 8.51 kcal·mol-1 for free DhaA and was increased to 9.55 kcal·mol-1 for the immobilized one. However, the solvent-accessible surface area ( m) increased from 3.69 to 4.00 kcal·(mol·mol·L-1)-1 after immobilization, which might be caused by the convenient moving of urea molecules into MCF-NH2 with electrostatic attraction. The amino and hydroxyl groups in the channel of MCF-NH2 could enhance DhaA rigidity through hydrogen bonding effectively reduced the effect of increasing of urea-accessible surface area, and improved the urea tolerance of DhaA. When DhaA was induced by DMSO, the Δ G(H2O) values of process from fold state to intermediate state were always 12.12 kcal·mol-1 in the before and after immobilization. The solvent-accessible surface area of DhaA dropped from 3.39 to 2.30 kcal·(mol·mol·L-1)-1 after immobilization, which might be caused by the block effect of amino and hydroxyl groups in MCF-NH2which effectively inhibiting the entry of non-polar DMSO molecules. From intermediate state to unfold state, the exposure of hydrophobic amino acids in DhaA leaded to an increase of m value, but the m value of immobilized DhaA [4.40 kcal·(mol·mol·L-1)-1] was still lower than the free DhaA [4.94 kcal·(mol·mol·L-1)-1] due to the polar microenvironment in the channel. Studying the unfolding process and thermodynamic parameters by fluorescence spectrometer is an effective technological mean for the research on DhaA stability, which can also offer a methodological guidance for stabilization mechanism study for other enzymes.
DhaA属于烷基卤脱卤酶(EC3.8.1.5, Haloalkane dehalogenases, HLDs), 能够有效降解1-氯丁烷、 1, 3-二氯丙烷、 1, 2, 3-三氯丙烷以及含其他卤素原子的农用杀虫剂[1]。 2001年, DhaA被首次发现对化学毒剂芥子气(HD)具有较高的催化反应活性, 之后迅速成为了国内外相关领域的研究热点[2, 3, 4, 5, 6]。 但是, DhaA在实际应用中稳定性不高, 特别是在尿素、 DMSO等变性剂影响下易丧失催化反应能力[7]。 固定化作为一种通用的生物酶稳定化技术, 能够通过载体与生物酶之间的相互作用提高生物酶的稳定性[8, 9]。 其中, 介孔泡沫(mesocellular foam, MCF)是一种三维笼状硅基介孔材料, 能够利用独特的孔径结构对生物酶进行固定化; 而经过氨基改性后, MCF还能够通过氢键作用进一步提高生物酶的稳定性, 但目前大部分研究只关注了MCF固定化后生物酶的功能变化, 对MCF固定化生物酶的稳定化机理并不清楚[10, 11, 12]。
荧光光谱作为研究生物酶构象的有效工具, 能够测定不同变性环境下生物酶的荧光光谱强度及位移, 通过荧光光谱变化考察热力学稳定性也已经成为评价生物酶稳定化效果的重要手段[13, 14]。 Meng[15]采用PEG修饰增强 srcSH3蛋白稳定性, 通过srcSH3蛋白在盐酸胍中荧光光谱的研究, 发现修饰后的初始吉布斯自由能变(Δ G(H2O))提高了0.93 kcal· mol-1。 Srimathi[16]尝试对α -淀粉酶进行糖基化修饰, 发现OSP400修饰提高了淀粉酶在硫氰酸胍溶液中的稳定性, Δ G(H2O)比未修饰的α -淀粉酶提高了7.86 kJ· mol-1。 Foord[17]考察了添加剂对细胞色素c和糜蛋白酶抑制剂2的稳定化效果, 发现盐酸胍中添加2M甘氨酸后, 两种生物酶的Δ G(H2O)分别提高了17.28和7.74 kcal· mol-1。 本文利用荧光光谱法研究了游离态DhaA和氨基改性介孔泡沫(MCF-NH2)固定化的DhaA在尿素和DMSO两种变性剂诱导下的变性过程, 通过比较固定化前后DhaA热力学参数的变化, 确定了MCF-NH2固定化DhaA的稳定化机理。
Infinite F50型酶标仪(Tecan公司, 奥地利), F-4600型荧光光谱仪(Hitachi公司, 日本)。 三嵌段共聚物(P123, 平均分子量5 800, Aldrich公司); 正硅酸乙酯(TEOS, 分析纯, 天津市福晨化学试剂厂); 盐酸(HCl, 分析纯, 北京化工厂); 1, 3, 5-三甲苯(TMB, 分析纯, 成都格雷西亚化学技术有限公司); 3-氨丙基-三乙氧基硅烷(APTES, 97%, J& K Scientific公司); 磷酸一氢钠、 磷酸氢二钠均为国药分析纯; DhaA由实验室提供, 蛋白浓度1.0 mg· mL-1, PB缓冲液pH 6.5。
1.2.1 MCF-NH2制备
2.0 g模板剂P123和5.7 mL扩孔剂TMB混合溶于52 mL去离子水中, 混合下逐滴加入10 mL浓度12 mol· L-1的盐酸, 混合液40 ℃下搅拌至澄清, 缓慢加入4.6 mL TEOS, 40 ℃搅拌下放置24 h。 将混合液转入带聚四氟乙烯内衬的水热反应釜, 100 ℃烘箱中反应24 h。 收集产物过滤, 洗涤清除模板剂, 将滤渣在马弗炉中550 ℃下煅烧5 h。 取0.7 g煅烧产物, 分散至35 mL甲苯中, 剧烈搅拌下逐滴加入14 mL APTES, 110 ℃回流24 h, 收集沉淀, 分别用乙醇和水冲洗后, 自然干燥得到MCF-NH2。
1.2.2 MCF-NH2固定化DhaA
取10 mg MCF-NH2, 超声分散后加入1 mL浓度为1.0 mg· mL-1的DhaA, 25 ℃下吸附4 h, 12 000 r· min-1离心2 min后倒出上清, 离心沉淀中加入pH 6.5 PB缓冲液, 重复重悬离心三次, 得到MCF-NH2固定化的DhaA, 命名为DhaA@MCF-NH2。
1.3.1 DhaA稳定性测定
取200 μ L不同变性程度的游离态DhaA(或DhaA@MCF-NH2)变性溶液, 加入10 mmol· L-1双(2-氯乙基)醚, 37 ℃下反应1 h。 加入50 μ L体积比30%的硝酸终止反应, 随后加入55 μ L硫氰化汞和110 μ L硫酸铁铵。 12 000 r· min-1离心2 min后, 取200 μ L上清加入96孔板, 测定460 nm下的吸收强度, 计算氯离子生成量。 以不添加变性剂时游离态DhaA(或DhaA@MCF-NH2)的氯离子生成量为1, 计算不同变性程度下DhaA的活性残留率。
1.3.2 DhaA变性荧光光谱和荧光相图测定
(1)尿素诱导DhaA变性
取蛋白浓度50 μ g· mL-1的游离DhaA(或DhaA@MCF-NH2)加入试管, 加入不同浓度的尿素溶液, 保证体系尿素终浓度0~6 mol· L-1, 25 ℃下放置12 h。 取上述不同变性条件下的DhaA溶液1 mL, 加入石英比色皿, 放入F-4600型荧光光谱仪, 固定狭缝宽度5.0 nm, 激发波长280 nm, 扫描范围300~400 nm, 扫描速率1 000 nm· min-1, 测定荧光发射光谱, 读取320和365 nm波长下的荧光强度(I320和I365), 扣除空白对照的荧光强度后, 绘制荧光相图。 空白对照中不含DhaA, 其余组分与上述变性溶液相同。
(2)DMSO诱导DhaA变性
变性及荧光测定方法相同, 区别在于变性体系DMSO终浓度为0~12 mol· L-1。
1.3.3 DhaA活性残留率与热力学参数计算
(1)二态模型
根据文献[18]报道的关于活性残留率与热力学参数的关系, 当DhaA在变性剂诱导下的变性过程符合二态模型时, 体系中DhaA仅存在折叠态(Folding, F)和去折叠态(Unfolding, U)两种状态。 这种情况下, DhaA分子从F态到U态的转变过程可以表示为

式中, KU为变性剂诱导下F态到U态的热力学平衡常数。
此时DhaA活性残留率与初始吉布斯自由能变符合式(2)
式(2)中, n, Δ G(H2O)和m分别为结合的变性剂分子数、 DhaA初始吉布斯自由能变以及单位浓度变性剂引发的自由能变。 将测定的r和[D]带入公式, 拟合后可得DhaA热力学参数Δ G(H2O)和m。
(2)三态模型
当DhaA的变性过程中存在折叠态(F)、 中间态(Intermediate, I)以及去折叠态(U)时, DhaA分子从F态到U态的转变过程可以表示为

式(3)中, KD1和KD2分别为变性剂诱导下F态到I态、 I态到U态的热力学平衡常数。 DhaA活性残留率与各阶段Δ G(H2O)的关系需要分开讨论:
①DhaA从F态向I态的转变阶段, r与Δ G(H2O)符合式(4)
②DhaA从I态向U态的转变阶段, r与Δ G(H2O)符合式(5)
将测定的r和[D]带入公式, 拟合后可得各变性阶段的热力学参数。
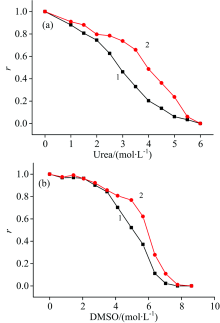
尿素和DMSO诱导DhaA变性过程中, DhaA残留活性随变性剂浓度的变化如图1所示。 可以看到, 随着尿素和DMSO浓度增加, 游离态DhaA活性逐渐降低, 当尿素浓度和DMSO浓度分别达到6和8 mol· L-1时, DhaA完全失活。 DhaA@MCF-NH2的活性同样随变性剂浓度增加而降低, 但降低幅度明显小于游离态DhaA, 说明在相同变性条件下DhaA@MCF-NH2比游离态DhaA活性更高。 在变性剂浓度达到某个临界值之前(尿素浓度为5.5 mol· L-1, DMSO浓度为7 mol· L-1), MCF-NH2固定化能够有效提高DhaA的稳定性; 而变性剂浓度过高时, 固定化DhaA迅速失活。 可能是由于变性剂分子大量存在的情况下, 影响了DhaA在MCF-NH2中的固定化, 导致MCF-NH2无法继续稳定化DhaA。
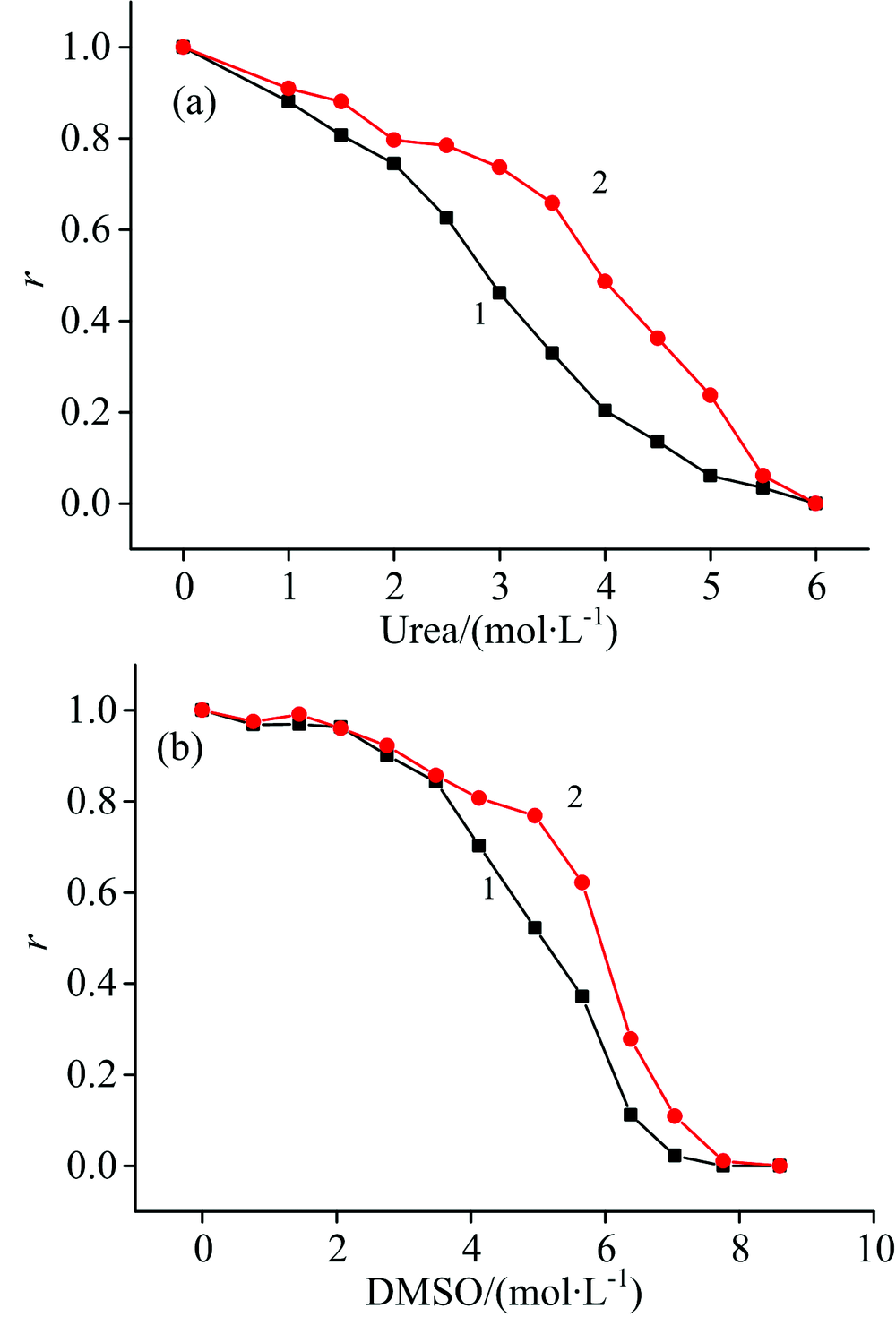 | 图1 不同浓度(a)尿素和(b)DMSO诱导变性条件下DhaA活性残留率 1: 游离态DhaA; 2: DhaA@MCF-NH2Fig.1 Residual activity ratio of DhaA at different concentrations of (a) urea and (b) DMSO in denaturation condition 1: Free DhaA; 2: DhaA@MCF-NH2 |
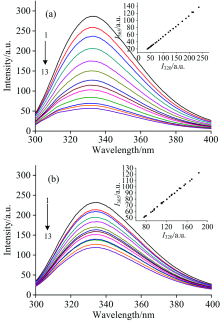
图2(a)和(b)分别是游离态DhaA和DhaA@MCF-NH2在不同浓度尿素中的荧光光谱图。 两种状态下DhaA的荧光强度均随尿素浓度的增加而逐渐降低, 最大发射波长位于332 nm左右, 在变性过程中没有出现明显位移。 DhaA@MCF-NH2荧光强度的衰减幅度明显低于游离态DhaA, 说明MCF-NH2固定化能够减小尿素对DhaA的影响。 在尿素诱导变性的荧光相图[图2(a)和(b)插图]中, 游离态DhaA和DhaA@MCF-NH2的I365/I320均保持线性变化, 说明DhaA在尿素诱导变性过程中不存在中间态, 变性过程符合Lumry-Eyring二态模型。
 | 图2 尿素诱导(a)游离态DhaA和(b) DhaA@MCF-NH2变性的荧光光谱(插图: 尿素诱导DhaA荧光相图) 1— 13: 体系中尿素浓度分别为0~6 mol· L-1, 间隔0.5 mol· L-1Fig.2 Fluorescence emission spectra representing the denaturation of (a) free DhaA and (b) DhaA@MCF-NH2 induced by urea (Insert: Phase diagram of fluorescence representing the denaturation of DhaA induced by urea 1— 13: The concentration of urea at 0~6 mol· L-1, interval concentration is 0.5 mol· L-1 |
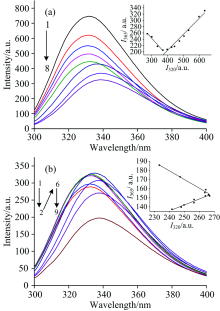
DMSO诱导DhaA变性的荧光光谱如图3所示。 游离态DhaA的荧光强度随着DMSO浓度的增加而显著降低[图3(a)中1— 5], 并且在DMSO浓度达到5.6 mol· L-1时, 荧光光谱开始发生红移, 说明此时DhaA构象开始发生改变[图3(a)中6— 8]。 由于MCF-NH2固定化的影响, DhaA@MCF-NH2荧光光谱的变化有所区别: DMSO浓度从0增大到1.4 mol· L-1时, DhaA@MCF-NH2荧光强度降低[图3(b)中1— 2]; DMSO浓度在1.4 mol· L-1增大到5.6 mol· L-1的过程中, DhaA@MCF-NH2荧光强度增大[图3(b)中3— 5]; 而DMSO浓度超过5.6 mol· L-1时, DhaA@MCF-NH2的荧光光谱开始发生红移[图3(b)中6— 9]。 游离DhaA和DhaA@MCF-NH2的荧光相图均呈两条直线[图3(a)和(b)插图], 证明DhaA在DMSO诱导变性过程中存在一个中间态, 两直线交点对应DMSO浓度5.6 mol· L-1, 与荧光光谱结果一致。 也就是说DMSO对DhaA的诱导变性分两个阶段: 当浓度从0增加到5.6 mol· L-1时, DhaA从折叠态(F)向中间态(I)转化; 当DMSO浓度继续增加时, DMSO诱导DhaA分子构象发生改变, DhaA从中间态(I)向去折叠态(U)转变, 变性过程符合三态模型。
 | 图3 DMSO诱导(a)游离态DhaA和(b) DhaA@MCF-NH2变性的荧光光谱(插图: DMSO诱导DhaA荧光相图) 1— 9: 体系中DMSO浓度分别为0~11.2 mol· L-1, 间隔1.4 mol· L-1Fig.3 Fluorescence emission spectra representing the denaturation of (a) free DhaA and (b) DhaA@MCF-NH2 induced by DMSO (Insert: Phase diagram of fluorescence representing the denaturation of DhaA induced by DMSO 1— 9: The concentration of DMSO at 0~11.2 mol· L-1, interval concentration is 1.4 mol· L-1 |
在两种变性剂诱导条件下, 分别采用式(2)、 式(4)和式(5)拟合计算得到的DhaA热力学参数如表1所示。 可以看到, 在尿素诱导变性情况下, 游离态DhaA的初始吉布斯自由能变Δ G(H2O)为8.51 kcal· mol-1, 单位浓度变性剂引发的自由能变m为3.69 kcal· (mol· mol· L-1)-1; DhaA@MCF-NH2的Δ G(H2O)为9.55 kcal· mol-1, 比游离态DhaA提高12.22%, m也提高到4.00 kcal· (mol· mol· L-1)-1。 固定化后m值升高, 证明DhaA的溶液可及面积有所增加, 即尿素分子更容易接近DhaA。 这是由于pH 6.5条件下, 尿素分子的氨基容易质子化, 而MCF-NH2孔道内表面残留羟基带负电荷, 尿素分子可以通过静电作用进入孔道, 引起m值的增加。 固定化后Δ G(H2O)增加, 这是因为MCF-NH2孔道内的氨基、 羟基能够通过氢键作用有效增强了DhaA的刚性。 比较DhaA的活性残留率, 可以发现MCF-NH2固定化带来DhaA刚性的增强有效降低了尿素可及面积增加带来的影响, 提高了DhaA的稳定性。
DhaA在DMSO诱导变性过程中符合三态模型, 当DhaA从F态向I态转化时, 游离态DhaA和DhaA@MCF-NH2的Δ G(H2O)基本一致, 但MCF-NH2固定化使得m从3.39 kcal· (mol· mol· L-1)-1降低为2.30 kcal· (mol· mol· L-1)-1, 下降率32.15%。 这是因为DMSO侧链为非极性甲基, MCF-NH2孔道内的极性基团有效阻碍了DMSO分子的进入, 减小了孔道中DhaA的溶液可及面积。 当DMSO浓度超过5.6 mol· L-1, DhaA从I态向U态转化时, DhaA开始出现构象变化, 内部疏水基团的暴露导致m增加, 但由于MCF-NH2孔道内极性基团的阻碍作用, 使DhaA@MCF-NH2的m值[4.40 kcal· (mol· mol· L-1)-1]仍然低于游离态DhaA[4.94 kcal· (mol· mol· L-1)-1]。
| 表1 游离态DhaA和DhaA@MCF-NH2的热力学参数 Table 1 The thermodynamic parameters of free DhaA and the DhaA@MCF-NH2 |
由此可见, 在DhaA变性过程中, MCF-NH2固定化技术能够改变DhaA的热力学参数。 在尿素诱导下, MCF-NH2固定化能够提高DhaA的初始吉布斯自由能变Δ G(H2O); 而DMSO诱导下, MCF-NH2固定化能够有效减小DhaA的溶液可及面积m, 两种变性条件下稳定化机理有所区别。
应用荧光光谱法对DhaA在尿素和DMSO两种变性剂诱导下的变性过程进行了研究, 进而就MCF-NH2固定化对DhaA的稳定化机理进行了分析。 通过研究发现, DhaA在尿素和DMSO诱导下的变性过程分别符合“ 二态模型” 和“ 三态模型” , MCF-NH2固定化不改变DhaA的变性过程, 但能够通过改变DhaA的热力学参数, 提高DhaA的稳定性。 其中, MCF-NH2固定化提高了DhaA在尿素中的初始吉布斯自由能变Δ G(H2O), 而在DMSO中减小了DhaA的溶液可及面积m。 利用荧光光谱法考察变性过程中生物酶的构象变化, 能够从热力学稳定性角度对固定化效果进行评价和分析, 但生物酶稳定化机理的深入解析, 还需要结合变性过程的动力学分析进行系统研究。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|