




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
咖啡酸分子表面增强拉曼光谱的理论与实验研究
[陈善俊1, *  , 范建
, 范建1 , 罗智能1 , 陈艳1, 2 , 李松1 , 张伟斌1 , 卢念1 , 韦建军3 ]
, 范建|
|


作者简介: 陈善俊, 1982年生, 长江大学物理与光电工程学院副教授 e-mail: chenshanjun2002@126.com
咖啡酸(CA)是一种具有很高的医学价值的药物成分, 在抗菌抗病毒方面应用广泛, 尤其是咖啡酸及其衍生物在抗肿瘤方面有着巨大作用, 现在对咖啡酸的相关研究越来越多, 但大部分都是关于咖啡酸医学性质的研究, 所以对咖啡酸分子的微观结构研究是非常有必要的。 目前关于CA在Ag表面上的表面增强拉曼散射(SERS)光谱的理论与实验结合的研究尚未见报道, 而对其振动光谱及表面增强机理的研究可以为咖啡酸的各种药学机理的研究提供一种科学的物理解释, 所以有必要将密度泛函理论(DFT)方法与表面增强拉曼散射技术相结合, 对咖啡酸在Ag纳米颗粒上的吸附性质及表面增强机理进行全面的研究, 这对推进它们在医药学等领域的相关研究有着重要的参考价值。 采用SERS与DFT技术对CA分子在Ag纳米颗粒表面上的表面增强拉曼光谱进行了研究。 在实验方面, 利用热还原反应原理, 使用柠檬酸三钠和硝酸银在加热搅拌情况下制备Ag纳米颗粒, 并使用激光共聚焦显微拉曼光谱仪测量了CA分子的常规拉曼散射(NRS)光谱及其表面增强拉曼散射(SERS)光谱。 在理论计算方面, 采用DFT的B3LYP方法, 以6-31+G**和LANL2DZ分别作为C, H, O和Ag的计算基组来优化咖啡酸的分子构型, 羟基与Ag4的吸附构型, 羧基与Ag4的吸附构型, 羟基与羧基共同与Ag4吸附的构型, 并以此为基础分析计算了CA分子的NRS光谱以及三种可能吸附模型的SERS光谱, 并结合实验结果进行比较。 同时对CA分子的振动模式进行了详细指认。 根据实验数据和理论结果分析, 在452 cm-1处的谱峰归属为环面外弯曲振动和O—H面外弯曲振动的耦合, 这说明CA分子上的酚羟基是与Ag纳米颗粒表面作用的, 不过相互作用较弱, 推测CA分子平面可能与Ag基底表面不垂直; 出现在1 338 cm-1处的谱峰归属于COO—伸缩振动, 则可以说明CA分子上的羧基可能与Ag纳米颗粒垂直吸附。 结果表明, CA分子是以羧基和酚羟基为吸附位吸附在Ag纳米颗粒表面上的。 同时对CA分子的振动模式进行了详细指认。 该工作对推进咖啡酸在生物医药等领域进一步的应用将起到重要作用。
, FAN JianCaffeic acid (CA) is a medicinal component with high medical value. It is widely applied in antibacterial and antiviral applications. In particular, caffeic acid and its derivatives have a enormous function in antitumor. Nowadays, there are many researches about caffeic acid. However, most of them are about the medicinal properties of caffeic acid, as a result the investigation of the microstructure of caffeic acid molecules is necessary. So far, there are no theoretical and experimental studies about the surface-enhanced Raman scattering spectroscopy (SERS) of CA on Ag surface. It is worth noting that the research on the vibrational spectrum and surface enhancement mechanism of caffeic acid can be a variety of pharmaceutical mechanisms of caffeic acid. Therefore, a combine surface-enhanced Raman scattering (SERS) and density functional theory (DFT) techniques are applied to conduct a comprehensive study of the adsorption properties and surface enhancement mechanism of caffeic acid on Ag nanoparticles, which can provide a Scientific explanation to the medicinal properties of caffeic acid. This has important reference for advancing their related research in medicine and other fields. In this paper, surface-enhanced Raman spectroscopy of CA molecules on the surface of Ag nanoparticles was studied using combined SERS and DFT techniques. Ag nanoparticles were prepared using trisodium citrate and silver nitrate under heating and stirring using the principle of thermal reduction reaction and conventional Raman scattering (NRS) spectra and SERS spectra of CA molecules were measured usingthe laser confocal micro-Raman spectrometer. In terms of theoretical calculations, we applied B3LYP method to optimize the molecular configuration of caffeic acid, the adsorption configuration of Ag4, the adsorption configuration of carboxyl group and Ag4, and the configuration of adsorption of Ag4 by both hydroxyl and carboxyl groups, using 6-31+G** and LANL2DZ as the basis set for C, H, O, and Ag, respectively. Then, the NRS spectra of CA molecules and the SERS spectra of three possible adsorption models were calculated and compared with experimental results. At the same time, the vibration mode of CA molecules was confirmed. According to the experimental data and theoretical results, the peak at 452 cm-1 was attributed to the coupling of the torsional bending vibration and the —OH out-of-plane bending vibration, which indicated that the phenolic hydroxyl group on the CA molecule have a weak interaction with the Ag nanoparticle. We speculated that the CA molecular plane may not be perpendicular to the surface of the Ag substrate. The peak appearing at 1 338 cm-1 was attributed to COO— stretching vibration, which indicated that the carboxyl group on the CA molecular is vertically adsorbed with the Ag nanoparticle. The results showed that CA molecules adsorbed on the surface of Ag nanoparticles with carboxyl groups and phenolic hydroxyl groups as adsorption sites. At the same time, we have identified the vibrational modes of CA molecules in detail. This work has an important effect on the further applications of caffeic acid in biomedicine and other fields.
咖啡酸(caffeic acid, CA), 化学式为C9H8O4, 是多种医学植物所含有的活性成分, 在蒲公英、 金银花、 仙人草等中含量较高, 有很好的医学药用价值, 特别是在抗菌抗病毒方面应用广泛[1]。 近年来由于对CA分子药理性质的广泛研究, 在灵敏度和稳定性检测方面提出了新的要求。 发生于金属良导体表面或特殊溶胶中, 能够产生高强度拉曼散射光谱的现象, 称为表面增强拉曼散射(surface-enhanced Raman scattering, SERS)[2], 可实现高灵敏度检测。 单分子SERS的实验突破及计算机的发展为SERS的理论研究提供了巨大的机遇。 拉曼光谱的计算是一个引人注目的研究方向, 为光谱分析识别谱峰提供理论支撑。 同时, 采用对比分析法比较理论值与实验值, 可以找到一些实验现象背后的理论解释。
密度泛函理论(density functional theory, DFT)是利用电子密度表示电子能量来描述多电子体系的量子理论, 可以对大多数多电子体系做到精确求解, 在物理、 化学等领域得到广泛应用[3]。 它考虑了电子相关作用, 有计算精度高、 理论可靠、 计算资源少等优点, 已被广泛应用于计算各种分子的结构和振动光谱[4, 5, 6]。 近年来, 采用DFT和拉曼技术研究咖啡酸(CA)的分子构型和振动光谱也成为研究热点。 Sá nchez-Corté s[7]等从实验上讨论了不同激发波长下咖啡酸在银溶胶中的表面增强拉曼光谱, Rincó n D[8]等采用从头计算和密度泛函理论讨论了咖啡酸酰胺的分子构型, Wagner[9]等研究了咖啡酸的常规拉曼光谱以及其吸附在TiO2颗粒上的振动光谱, 刘靖丽[10]等采用DFT方法研究了CA分子的能量最低稳定构型和其红外光谱及拉曼光谱。 然而, 目前关于CA在Ag表面上的SERS光谱的理论与实验结合的研究尚未见报道。 鉴于咖啡酸在医学上的药理作用及目前的研究现状, 我们有必要结合SERS和DFT技术, 对咖啡酸在Ag纳米颗粒上的吸附性质及表面增强机理进行全面的研究, 这对推进它们在医学, 药理学等领域的相关研究有着重要的参考价值。
本文理论计算部分采用Gaussian 09程序包[11], 运用DFT中常用的B3LYP方法[12]对咖啡酸分子以及其与银纳米颗粒吸附的模型进行优化, 将CA分子NRS光谱和SERS光谱的理论结果与实验测量的结果进行分析对比, 从吸附位、 吸附构型等方面研究咖啡酸分子在Ag纳米颗粒上的吸附机理, 并借助Gauss View5.0可视化软件对拉曼谱峰的振动模式进行了详细指认。
柠檬酸三钠(Na3C6H5O7· 2H2O)为优级纯, 其含量≥ 99.5%; 咖啡酸(实验试剂)含量≥ 98%, 纯度为GR; 硝酸银(AgNO3)含量≥ 99.5%, pH值5.0~6.0, 纯度为AR。 所有化学试剂由中国Sinopharm Chemical Reagent(国药化试)公司生产。 在制备银溶胶时, 把三次蒸馏去离子水作为实验溶剂, 提纯银溶胶样品采用德国Sigma公司, 规格为SIGMA 3-18k, 最大转速可达18 000 r· min-1的高速台式冷冻型离心机; 使用由日立公司生产的型号为H-800的透射电子显微镜表征银溶胶的微观形貌; 测拉曼光谱时, 采用英国Renishaw公司2003年生产的, 光谱范围为200~1 000 nm(我们选取波长为750 nm的激光光源), 最低波数为10 cm-1, 型号为inVia的激光共聚焦显微拉曼光谱仪。
关于银溶胶的制备, 其中比较简单实用的就是Lee-Meisel[13]法, 利用柠檬酸钠还原硝酸银(AgNO3)。 取250 mL去离子水, 再将45 mg的AgNO3溶解在溶液中, 使用恒温加热搅拌器加热至沸腾, 同时逐渐将5 mL的Na3C6H5O7· 2H2O(浓度为1%)用胶头滴管滴入AgNO3溶液中, 在沸腾10 min左右停止加热, 待冷却到室温。 可以观察到溶液颜色变成了灰褐色或者灰绿色, 此时制备的银胶体颗粒直径符合实验的要求。 银胶体颗粒的TEM图如图1所示。
 | 图1 银基底的TEM图像Fig.1 TEM image of silver substrate |
本文利用DFT中的B3LYP[12]方法完成理论计算, 用6-31+G* * 基组计算氧, 碳和氢原子, 用LANL2DZ赝势基组计算银原子, 所有计算在Gaussian 09软件上进行。 使用Gauss View 5.0软件建立咖啡酸(CA)分子吸附在银基底表面的可能吸附模型, 并对其构型进行优化, 使其处于能量最低稳定态。 在得到稳定结构的基础上, 用同样的方法和基组计算CA分子的SERS光谱。 最后, 为对CA分子振动基频进行精确归属指认, 借助了GaussView5.0可视化软件来完成。
在构造合适的吸附构型时, CA分子和Ag纳米颗粒的表面特性对吸附分子在基底表面的吸附行为有着非常大的影响。 如果不建立简化模型, 对于溶液里如此复杂庞大的体系是无法精确计算的, 因此, 考虑到Ag原子(原子序数为108)有较大的质量, 在进行理论模拟时, 可以用Ag团簇来代替银纳米颗粒作为基底; 而且与固体银晶体里相互之间连接紧密的银原子相比, Ag纳米颗粒里的银原子更为活泼, 使得CA分子所吸附的银原子相对容易松动。 基于以上两点, 可以采用简化模型模拟CA分子与Ag纳米颗粒的吸附行为。
应用DFT方法对CA分子进行结构优化, 得到不存在虚频的分子结构, 表明优化的分子结构处于能量最低稳定状态。 优化得到的CA分子构型如图2所示。
 | 图2 咖啡酸分子的球棍模型Fig.2 The ball-and-stick model of caffeic acid |
为了确定CA分子与Ag纳米颗粒的吸附方式, 本文构建了三种可能吸附构型, 优化后的吸附构型如图3所示。 模型A是CA分子的羧基与Ag4团簇作用; 模型B是CA分子的酚羟基与Ag4团簇作用; 模型C为CA分子的羧基和酚羟基共同吸附在Ag4团簇上。
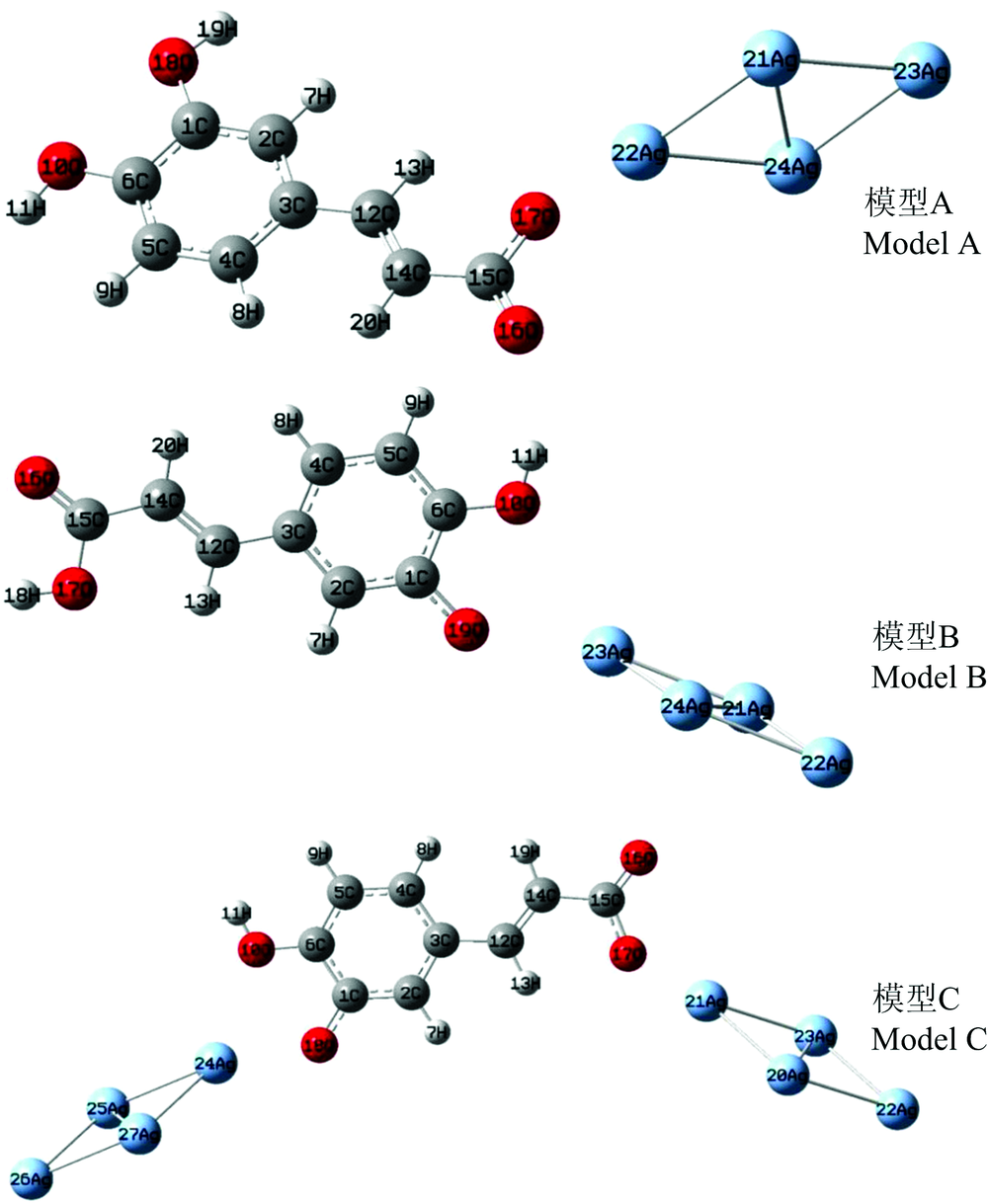 | 图3 CA分子在Ag胶体表面的吸附构型Fig.3 Adsorption models of CA molecules on Ag colloid surface |
CA的常规拉曼(normal Raman scattering, NRS)和表面增强拉曼(SERS)的实验结果如图4所示, 其理论计算结果如图5所示。 并且参考了相关文献[8, 14]对CA分子的振动频率及其振动模式进行了详细的指认, 详见表1。 在表1中, 可发现一种振动频率可能是对应几种振动模式的耦合, 这是由CA分子的对称性低及其侧链结构所致。
 | 图4 CA粉末的NRS光谱(a)和CA在银溶液中的SERS光谱(b)实验结果Fig.4 Experimental results of the NRS spectrum of CA powder (a) and the SERS spectrum of CA in silver solution (b) |
 | 图5 CA分子拉曼光谱理论计算对比曲线图 a: 常规; b: 羧基吸附; c: 酚羟基吸附; d: 羧基和酚羟基共同吸附Fig.5 Contrast curves of CA molecular Raman spectroscopy a: Normal; b: Carboxyl adsorption; c: Phenol hydroxyl adsorption; d: Co-adsorption of carboxyl and phenolic hydroxyl |
| 表1 CA分子理论模拟结果和实验观察结果的振动模式指认表 Table 1 Calculated, observed and assignment of the vibrational frequencies (cm-1) for caffeic acid and caffeic acid adsorbed on Ag surface (CA-Ag) |
对图4中CA分子的实验SERS光谱(b)指认分析。 谱带在452 cm-1出现的振动峰, 借助于Gauss View 5.0可视化软件将其归属于环面外弯曲振动和O— H面外弯曲振动的耦合, 由此可以说明CA分子上的酚羟基是与Ag纳米颗粒表面作用的, 其振动强度较弱, 则表明O— H键与基底表面可能不垂直。 出现在1 338 cm-1的较强谱峰归属于COO— 伸缩振动, 则可以说明CA分子上的羧基可能与Ag纳米颗粒垂直吸附。 归属于苯环上C— C伸缩振动的1 120 cm-1峰和1 435 cm-1峰, 都存在着较强的振动强度, 根据表面增强拉曼的选择定则[15], 可以判断基底表面与CA分子平面垂直或接近垂直。 综上所述, 表明模型C更符合实际情况, 即CA分子是通过羧基和酚羟基共同吸附在基底表面的, 且苯环平面与基底表面可能垂直或接近垂直。
比较分析图5和表1, 可以发现, 被指认环面外弯曲振动和COO— 面内弯曲振动的耦合的608 cm-1峰, 出现在1 120 cm-1的O— H面内弯曲振动, C— C伸缩振动和C— H弯曲振动的耦合, 对应C— O伸缩振动, C— H和O— H面内弯曲振动耦合的1 275 cm-1峰, 以及在1 332 cm-1的C— H面内弯曲振动和— COO— 伸缩振动耦合都与实验结果符合得非常好。 在理论模拟结果中, 出现在937 cm-1的C— H面外弯曲振动和出现在1 651 cm-1的C— H面内弯曲振动, 以及归属于C═O对称伸缩振动的1 678 cm-1峰与实验值存在着30~50 cm-1之间的误差。 该分析结果进一步说明模型C的合理性。 即CA分子在银胶体溶液中是通过羧基和酚羟基共同吸附在Ag纳米颗粒表面上的, CA分子平面与Ag纳米颗粒表面呈垂直或接近垂直状态。
基于表面增强拉曼散射和密度泛函理论, 借助Gaussian09软件对咖啡酸分子吸附在Ag纳米颗粒上的吸附体系进行了结构优化及其拉曼光谱计算, 通过对NRS和SERS理论和实验结果的对比分析, 探究了咖啡酸分子在Ag溶胶中的吸附性质, 并进一步详细指认归属了它的振动基频。 通过分析与羧基、 羟基取向有关的结构变量, 建立了三种不同的可能吸附构型, 并与相关的实验值进行比较。 结果表明, 基于模型C的理论结果与相应实验结果符合得更好, 为此可确定, 在Ag溶胶中, CA分子侧链的羧基和苯环上的羟基都与Ag纳米颗粒相互作用, 而且整个苯环平面可能与基底面垂直。 该研究结果对CA分子进一步的微观结构及其药理性质的研究具有重要的参考意义。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|