引用本文
[J]. 光谱学与光谱分析, 2019,39(3): 977-981.
C. Charanya, S. Sampathkrishnan, N. Balamurugan. Natural Bond Orbital (NBO), Natural Population Analysis and Mulliken Analysis of Atomic Charges of 4-Amino-3-Phenylbutanoic Acid[J]. Spectroscopy and Spectral Analysis, 2019,39(3): 977-981.
Doi:10.3964/j.issn.1000-0593(2019)03-0977-05
Permissions
C. Charanya, S. Sampathkrishnan, N. Balamurugan. Natural Bond Orbital (NBO), Natural Population Analysis and Mulliken Analysis of Atomic Charges of 4-Amino-3-Phenylbutanoic Acid[J]. Spectroscopy and Spectral Analysis, 2019,39(3): 977-981.
Doi:10.3964/j.issn.1000-0593(2019)03-0977-05
Copyright©2019, 《光谱学与光谱分析》期刊社
《光谱学与光谱分析》期刊社 所有
Natural Bond Orbital (NBO), Natural Population Analysis and Mulliken Analysis of Atomic Charges of 4-Amino-3-Phenylbutanoic Acid

Abstract
The Molecular Structure of 4-Amino-3-phenylbutanoic acid conformers have been studied in the gas phase. Natural Bond Orbital Analysis (NBO) and Mulliken analysis of atomic charges of 4-Amino-3-phenylbutanoic acid have been performed by DFT level of theory using B3LYP/6-311++G(d,p) basis set. The atomic charges, electronic exchange interaction and charge delocalization of the molecule have been performed by Natural Bond Orbital(NBO) analysis and Natural Population Analysis(NPA) have been constructed at B3LYP/6-311++G(d,p) level.
Keyword:
DFT; NBO; NPA
中图分类号:O56
文献标志码:A
Introduction
4-Amino-3-phenylbutanoic acid (Phenibut) is employed as an orally bio available agent for the treatment of Spinal Muscular Atrophy (SMA)[1]. Literature survey reveals that so far there is no Natural Bond Orbital Analysis and Mulliken Analysis of atomic charges study for the Phenibut compound. In this study, The NBO analysis is carried out to investigate in various intra-inter molecular interactions of molecular system and also provides a convenient basis for investigating charge transfer or conjugate interactions in molecular system. Some electron donor, acceptor orbital and the interacting stabilization energy resulted from the E(2) (Energy of hyper conjugation interaction) are reported. Moreover, the Mulliken Analysis of atomic charges of the Phenibut compound have been calculated and the calculated results have been reported.
1 Methods of Analysis
The Molecular structure of Phenibut along with numbering of atoms is shown in Fig.1. The Natural Bond Orbital calculations were carried out using Gaussian 09W program package[2], invoking gradient geometry optimization[3]. Initial geometry generated from standard geometrical parameter was minimized without any constraint in the potential energy surface in the standard 6-311++G(d, p) basis set. This geometry was then reoptimized at three parameter hybrid functional (B3) Lee-Yang-Parr (LYP) level using 6-311++G(d, p) basis set. The localized basis set completely describes the wave functions in the most economic method, as electron density and other properties are described by the minimal amount of filled Natural Bond Orbital’ s (NBO) which describe the hypothetical, strictly localized Lewis structure which can be used as the measure of delocalization. This non-covalent bonding and anti-bonding charge transfer interactions can be quantitatively described in terms of the second order perturbation interaction energy (E(2))[4, 5, 6, 7].
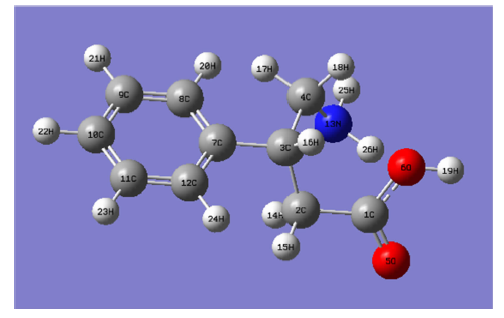 | Fig.1 The theoretical optimized geometric structure with atoms numbering of Phenibut |
{kind=link}
2 Result and Discussion
2.1 Natural Bond Orbital (NBO) Analysis
Natural Bond Orbital(NBO) analysis is one of the most powerful tools for interpreting quantum-chemical results in terms of chemically significant terms. This method localizes the molecular wave functions in optimized electron pairs, corresponding to lone pairs; core pairs on bonding units; giving a picture which is close to the familiar Lewis picture of molecular structure. In order to understand various second order interactions between the filled orbitals of one subsystem and vacant orbitals of another subsystem and vacant orbitals of another subsystem, the NBO calculations were performed using NBO 3.0 program[8] as implemented in the Gaussian 09W package at the DFT/B3LYP/6-311++G(d, p) level.
The Natural Bond Orbital (NBO) analysis provides an efficient method for studying intra and inter-molecular bonding and interaction among bonds, and also provides a convenient basis for investigating charge transfer or conjugative interaction in molecular system[9]. The second-order Fock-matrix was carried out to evaluate different types of donor-acceptor interactions and their stabilization energies in the NBO basis[10]. The interaction results are a loss of occupancy from the localized NBO of the idealized Lewis structure into an empty non-Lewis orbitals.
The NBO analysis has been performed to elucidate the intramolecular interaction, rehybridization and delocalization of electron density within the molecule. Which are presented in Table 1. In order to investigate the intramolecular interaction, the stabilization energies of the Phenibut compound were computed by using second-order perturbation theory. The results of second-order perturbation theory analysis of Fock matrix at the B3LYP levels with 6-311++G(d, p) basis set of the Phenibut compound are given in Table 1.
| Table 1 Second-order perturbation theory analysis of Fock matrix in NBO basis corresponding to the intramolecular of the Phenibut compound |
In the molecule, a strong intramolecular hyperconjugative interaction of π electron with the greater energy contributions from O6→ C1-O5 (49.66 kcal· mol-1 for the DFT/B3LYP level), C11-C12→ C7-C8 (48.58 kcal· mol-1 for the DFT/B3LYP level), C7-C8→ C9-C10 (46.75 kcal· mol-1 for the DFT/B3LYP level), O5→ C1-O6 (49.66 kcal· mol-1 for the DFT/B3LYP level) for the amino ring of the studied molecule.
In Table 2. C1-C2 orbitals with 1.979 electrons have 50.59% C1 character in a sp1.47 hybrid and have 49.41% C2 character in a sp2.99 hybrid. C1-O5 orbitals with 1.996 electrons has 34.06% C1 character in a sp2.08 hybrid and has 65.94% C2 character in a sp1.33 hybrid. C1-O6 orbitals with 1.995 electrons has 30.15% C1 character in a sp2.69 hybrid and has 69.85% C2 character in a sp1.55 hybrid. C1-N13 orbital with 1.994 electrons have 40.01% C1 character in a sp3.06 hybrid and has 59.99% C2 character in a sp2.08 hybrid.
| Table 2 NBO results showing formation of Lewis and non-Lewis orbitals for Phenibut |
2.2 Natural Population Analysis
The Natural population analysis[11, 12] performed on the Phenibut molecule clearly describes the distribution of charges in the various sub-shells (Core, Valence, Rydberg) in the molecular orbital. The accumulation of natural charges on individual atom of the Phenibut molecule is given in Table 2. It shows that an atoms C1 has the most electronegative charge of -5.0636e and O6 has the most electropositive charge of 8.7849e. Conversely, the N13 and C12 atoms have considerable electropositive and they are tending to acquire electron. Further, the natural population analysis showed that 96 electrons in the title molecule are distributed on the sub shell as follows:
Core: 25.99013 (99.9621% of 26)
Valence: 69.65012 (99.5002% of 70)
Rydberg: 0.35975 (0.3747% of 96)
2.3 Charge Distribution
The charge distribution of the molecule has calculated on the basis of Mulliken method[13] using B3LYP/6-311++G(d, p) level theory. This calculation depicts the charges of the every atom in the molecule. Distribution of positive and negative charges is the vital to increasing or decreasing of bond length between the atoms. The survey of literature reveals that effective atomic calculations gave an important role in the application of chemical calculation to molecular system because of atomic charges, dipole moment, molecular polarizability, electronic structure, acidity-basicity behavior and more lot of properties of molecular system[13]. Mulliken atomic charges and plot has shown in Table 4 and Fig.2 respectively. The Mulliken charge analysis of Phenibut shows that presence of two oxygen atoms (O5=-0.2343; O6=-0.1925 imposes positive charges on C3, C7 atoms. However, C3, C7 possess positive charge due to negative charge (-0.3557) of atom N13. Moreover, the positive charge distribution observed on the remaining hydrogen atoms (H14, H15, H16, H17, H18, H19, H20, H21, H22, H23, H24, H25 and H26).
| Table 3 Accumulation of natural charges and electron population of atoms in core, valance, Rydberg orbitals of Phenibut |
| Table 4 Mulliken’ s atomic charges of Phenibut at B3LYP/6-311++G(d, p) level of theory |
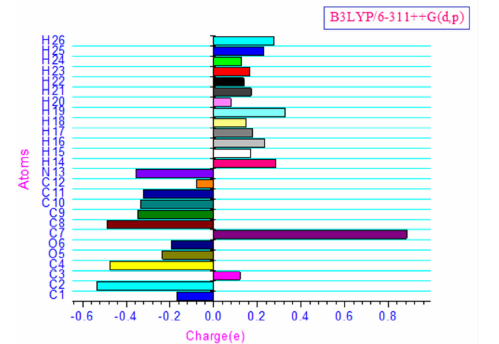 | Fig.2 The Historam of calculated Mulliken charge for phenibut |
{kind=link}
3 Conclusion
In the present work, the optimized molecular structure of the title compound have been calculated by DFT/B3LYP method with 6-311++G(d, p) level calculations. The NBO analysis provides an efficient method for studying inter and intra molecular interaction in molecular system. The stabilization energy has been calculated from second order perturbation theory. NBO theory provides an excellent approach to interpret the infrared spectra in electronic terms. Relative band positions, together with displacement of bands corresponding to molecular groups not involved in hydrogen bonds, are explained by this theory. In this paper, NBO analysis was also used to evaluate the influence of the intermolecular interactions by comparing the results obtained for the single point and optimized structures. The NBO analysis confirms the hyper conjugation interaction. The strengthening and increase in wave number is due to the substitution of OH and CH2 groups, respectively.
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|