引用本文
Rubarani. P. Gangadharan, S. Sampath Krishnan, M. Thirumalaikumar. Quantum Chemical and Corrosion Inhibition Studies of(4-Chlorophenyl)-N-(4-Methylphenyl) Nitrone[J]. Spectroscopy and Spectral Analysis, 2019,39(12): 3940-3945.
Doi:10.3964/j.issn.1000-0593(2019)12-3940-06
Permissions
Doi:10.3964/j.issn.1000-0593(2019)12-3940-06
Copyright©2019, 《光谱学与光谱分析》期刊社
《光谱学与光谱分析》期刊社 所有
Quantum Chemical and Corrosion Inhibition Studies of(4-Chlorophenyl)-N-(4-Methylphenyl) Nitrone

Abstract
The compound (4-chlorophenyl)-N-(4-methylphenyl) nitrone (4CPNMPN) has been selected as one of the new nitrone derivative for our study. The molecular structure of the compound was investigated based on frontier orbital analysis and natural bond orbital (NBO) theory. The present work also focuses on the inhibition efficiency of the compound. It is an attempt to find the correlation between the molecular structure of the compound and possible behaviour like corrosion inhibitors. The NBO analysis and the values of electric dipole moment ( μ) of the investigated molecule were computed using DFT calculations. The molecule orbital contributions were studied by using the total (TDOS) density of states. The strong evidences that the compound can be used as an efficient nonlinear optical (NLO) of 4CPNMPN were demonstrated by considerable polarizability and hyperpolarizability values obtained at DFT levels.
Keyword:
Molecular orbital analysis; Corrosion inhibition; NBO analysis

IntroductionNitrones were first prepared by Beckmann in 1890[1] and the name nitrone was derived from abbreviation of “ nitrogen-ketones” by Pfeiffer in 1916 to highlight their resemblance to ketones. Many nitrone derivatives are found to possess pharmacological activity[2] and form an essential part of the molecular structure of important drugs. Many diseases of ageing, including stroke, cancer development, Parkinson’ s disease and Alzheimer disease are known to have enhanced levels of free radicals and oxidative stress. Recent research has shown that alpha-phenyl-tertbutyl nitrone (PBN) related nitrones also have anti-cancer activity in several experimental cancer models and have potential as therapeutics in some cancers[3]. We have selected (4-chlorophenyl)-N-(4-methylphenyl) nitrone, abbreviated as 4CPNMPN, one of the new nitrone derivative for our study. To the best of our knowledge, despite of potential pharmacological applications, this molecule has not undergone any comprehensive stereoelectronic and spectroscopic studies. The present work reports a computational study and molecular structure of (4-chlorophenyl)-N-(4-methylphenyl) nitrone. The molecular orbital, natural bond orbital, corrosion inhibition and non linear optical behaviour have been analyzed.
1 Computational details
Computational aspects for geometry optimization and electronic structure of the compound have been done by density functional theory[4] by using the Gaussian 03W program package[5] employing different basis sets and Becke’ s three parameter hybrid exchange functionals with Lee-Yang-Parr correlation functionals (B3LYP)[6, 7, 8]. In order to investigate intra-molecular charge transfer interactions, rehybridization and delocalization of electron density within the molecule, the natural bonding orbitals (NBO) analysis has been performed. To calculate functional group contributions to the molecular orbitals, the total density of states (TDOS or DOS) spectrum was prepared by using the program GaussSum 2.2[9]. The contribution of a group to a molecular orbital was calculated by using Mulliken population analysis.
2 Results and Discussions
2.1 Geometry Optimization
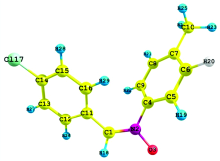
The theoretical structure of molecule having the molecular formula C14H12ClNO have been calculated by DFT using B3LYP functional having basis set 6-31++G (d, p) with the help of Gaussian 03W package and geometry obtained from B3LYP/6-31++G (d, p) is shown in Fig.1.
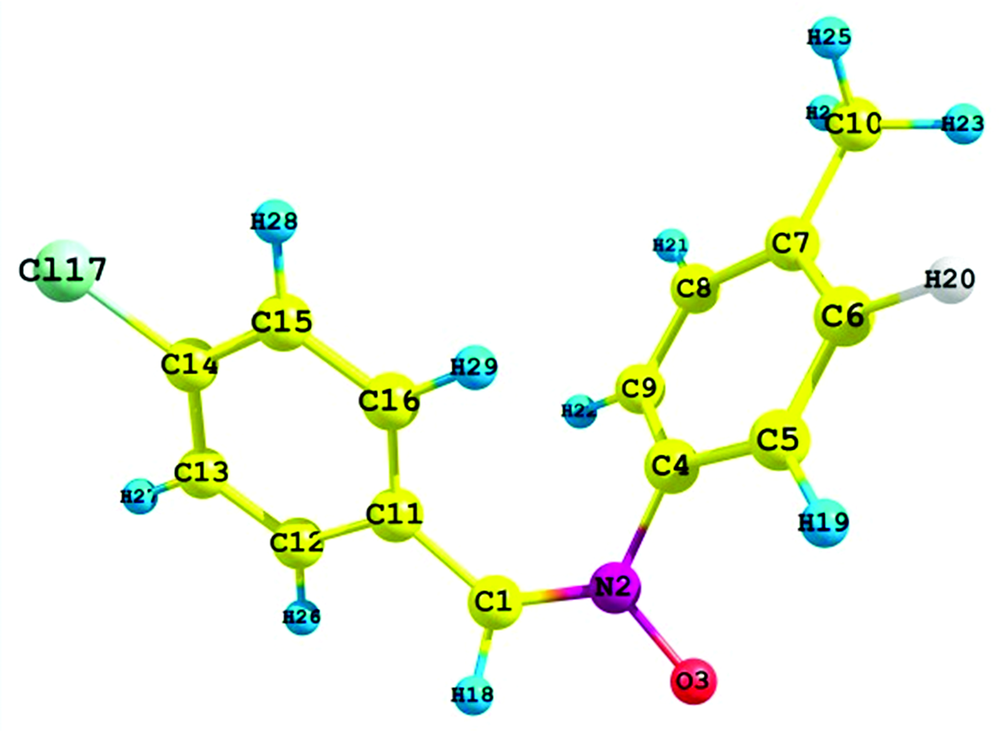 | Fig.1 Optimized molecular structure and atomic numbering of 4CPNMPN |
{kind=link}
2.2 Analysis of frontier molecular orbitals (FMOs)
The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are the main orbitals that plays an important role in chemical stability. The HOMO is the orbital that primarily acts as an electron donor and the LUMO is the orbital that largely acts as the electron acceptor[10]. The MOs are defined as eigen functions of the Fock operator, which exhibits the full symmetry of the nuclear point group, they necessarily form a basis for irreducible representations of full point-group symmetry. The HOMO and LUMO energy calculated by B3LYP/6-31++G (d, p) level of theory show the energy gap which reflects the chemical activity of the molecule. The pictorial illustration of the frontier molecular orbitals and their respective positive and negative regions are shown in Fig.2. Molecular orbitals is viewed in a qualitative graphical representation which can provide insight into the nature of reactivity, and some of the structural and physical properties of molecules. The positive and negative phase is represented in red and green colour, respectively. The region of HOMO level spread over the chlorine atom and the phenyl group. The region of LUMO level spread approximately over the entire molecule and the calculated energy gap of HOMO-LUMO’ s explains the ultimate charge transfer interface within the molecule[11].
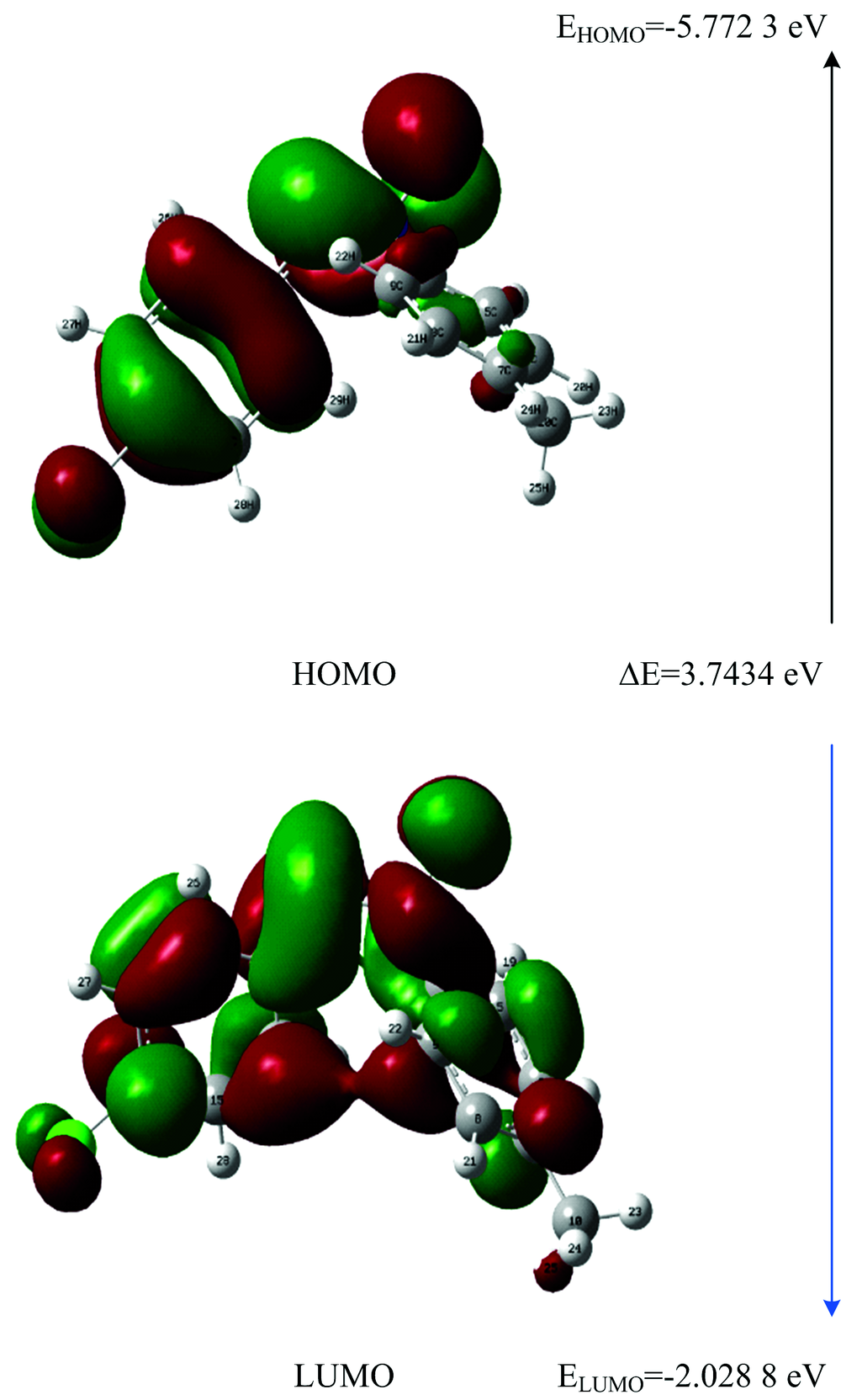 | Fig.2 Frontier molecular orbitals of 4CPNMPN at B3LYP/6-31++ G(d, p) level |
{kind=link}
HOMO energy (B3LYP)=-5.772 3 eV
LUMO energy (B3LYP)=-2.028 8 eV
HOMO-LUMO energy gap (B3LYP) Δ E=3.743 4 eV
On the basis of HOMO-LUMO energies global reactivity descriptors, such as the energies of frontier molecular orbitals (EHOMO, ELUMO), energy band gap (EHOMO-ELUMO), electronegativity (χ ), chemical potential (μ ), global hardness (η ), global softness (S) and global electrophilicity index (ω ), which describe the electrophilic behaviour[12, 13, 14, 15, 16] have been calculated for 4CPN4MPN using equations (1— 5). The chemical hardness and softness of a molecule is a good indicator of the chemical stability of a molecule[17]. From the HOMO-LUMO energy gap, one can find whether the molecule is hard or soft. The molecules having large energy gap are known as hard and molecules having a small energy gap are known as soft molecules. The soft molecules are more polarizable than the hard ones because they need small energy for excitation.
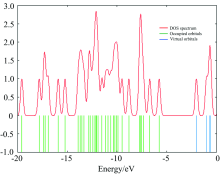
In this work Gauss-Sum 2.2 program has been used to calculate group contributions to the molecular orbital’ s and prepare the density of the state (DOS) as shown in Fig.3. The DOS spectra were created by convoluting the molecular orbital information with Gaussian curves of unit height. In addition, the decrease in the HOMO and LUMO energy gap explains the eventual charge transfer interaction taking place within the molecule which is responsible for the chemical activity of the molecule.
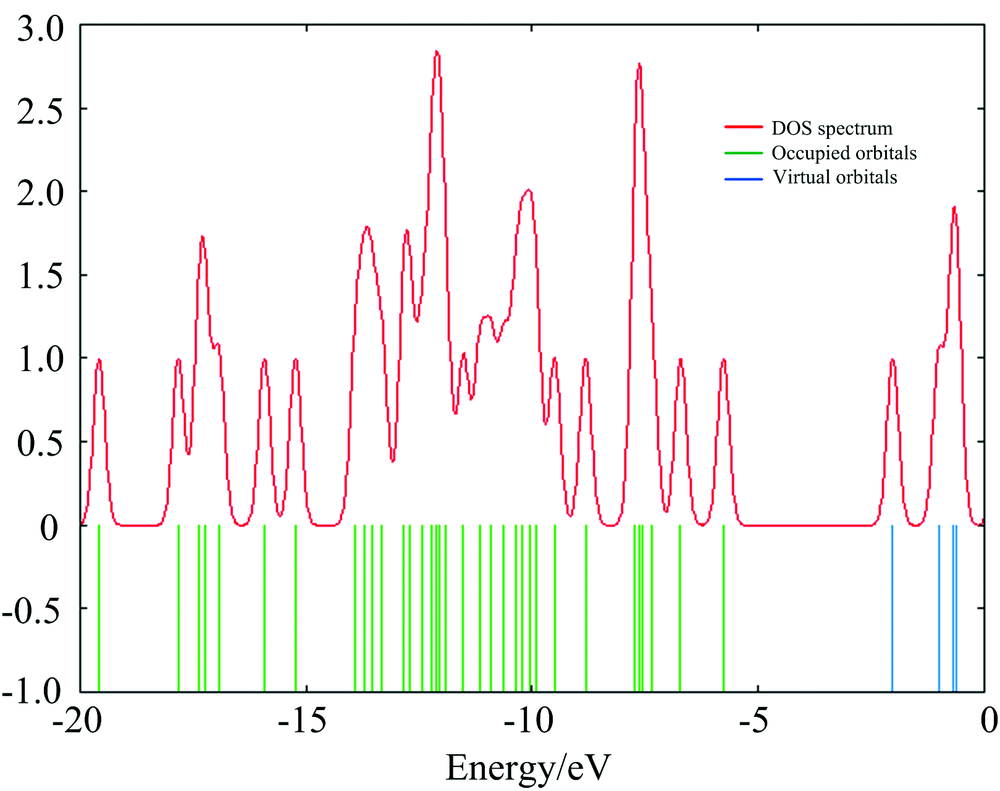 | Fig.3 The calculated DOS spectrum for 4CPNMPN |
{kind=link}
2.3 Corrosion inhibition studies
One of the most common effective and economic method to protect metals against corrosion is the use of organic compounds as corrosion inhibitors[18, 19]. The inhibition property of a compound has been often correlated with energy of HOMO, LUMO and HOMO-LUMO gap. A good correlation has been found between the speeds of corrosion and EHOMO that is often associated with the electron donation ability of the molecule. Survey of literature shows that the adsorption of the inhibitor on the metal surface can occur on the basis of donor-acceptor interaction between the π -electrons of the heterocyclic compound and the vacant d-orbitals of the metal surface atoms[20, 21], high value of EHOMO of the molecules show its tendency to donate electrons to appropriate acceptor molecules with low energy empty molecular orbitals. Increasing values of EHOMO facilitate adsorption and therefore enhance the inhibition efficiency, by influencing the transport process through the adsorbed layer. Similar relations were found between the rates of corrosion and Δ E[22, 23, 24]. The energy of the lowest unoccupied molecular orbital indicates the ability of the molecule to accept electron. The lower the value of ELUMO, the more probable the molecule would accept electron. Consequently, concerning the value of the energy gap Δ E, larger values of the energy difference will provide low reactivity to a chemical species. Lower of the Δ E will render good inhibition efficiency, because the energy required to remove an electron from the lowest unoccupied orbital will be low[25].
From the values of HOMO and LUMO it can be seen that higher values of EHOMO (EHOMO=-5.772 3 eV) indicates better inhibition efficiency. The binding ability of the inhibitor to the metal surface increases with increasing of the HOMO and decreasing of the LUMO energy values. In our studies, inhibitor having low value ELUMO=-2.028 8 eV) could have better performance as corrosion inhibitor. Lower values of the energy difference will render good inhibition efficiency, because the energy to remove an electron from the last occupied orbital will be low[26]. These results Δ E=3.743 4 eV has low energy gap, which shows that molecule could have better performance as corrosion inhibitor.
2.4 Natural Bond Orbital Analysis
NBO analysis is an efficient method for study of the intramolecular and intermolecular bonding and interactions among bonds. This analysis also provides the study of filled NBOs (donors) and empty NBOs (acceptors) and their interactions with the stabilization energy E(2) resulting from the second-order perturbation theory. The larger the E(2) value, the more intensive is the interaction between electron donors and acceptors, i.e. the more electron donating tendency from electron donors to acceptors and the greater the extent of conjugation of the whole system. This interaction results a loss of occupancy from the concentration of electron NBO of the idealized Lewis (bond or lone pair) structure into an empty (anti-bond or Rydberg) non-Lewis orbital.
The second-order perturbation theory analysis of Fock matrix in NBO basis of 4CPNMPN molecule display strong intra-molecular conjugative and hyperconjugative interactions and demystify the rehybridization and delocalization of electron density within the molecule. Some important interactions between Lewis and non-Lewis orbitals along with their interacting stabilization energies are shown in Table 2.
| Table 2 The dipole moments μ (D), polarizability (Δ α ), the average polarizability (α ) and the first hyperpolarizability (β tot) of 4CPNMPN calculated by B3LYP method |
The Fock matrix analysis shows strong intra-molecular hyperconjugative interactions of π electrons between π bond orbitals and antibonding orbitals. These interactions are established by the orbital overlapping between π (C— C or C— H) and π * (C— C or C— H) bond orbitals resulting ICT (Intramolecular charge transfer) causing stabilization of the system.
The electron density (ED) at the six conjugated π bonds (1.5~1.7 e) and π * antibonds (0.1~0.4 e) of the phenyl ring clearly shows strong delocalization leading to stabilization of energy in the range of 16~43 kcal· mol-1. These results are consistent with as reported by Reed[27]. The important interaction (n— π ) energies associated with the resonance in the molecule are electron donation from the LP (3) of atom O3 to the anti-bonding acceptors π * (C1-N2) which correspood to the stabilization energies 58.60 kcal· mol-1 respectively. A strong intramolecular interaction of π electrons occurs from π (C6-C7) and π (C11-C12) bonds to the π * (C4-C5) and π * (C13-C14) antibonds corresponding to the stabilization energies 23.45 and 24.57 kcal· mol-1 respectively. The most important interaction energy in this molecule is electron donating from O3 LP (2) to the antibonding acceptor σ * (C1-N2) resulting moderate stabilization energy of 7.72 kJ· mol-1. The maximum energy delocalization take part in the π — π * transition. These intramolecular charge transfer (n— σ * , n— π * and π — π * ) may induced biological activities in the molecume. The E(2) values and types of the transition is shown in Table 1.
| Table 1 Second order perturbation theory analysis of Fock matrix in NBO basis for 4CPNMPN |
2.5 Nonlinear Optical Properties and Dipole Moment
Density functional theory has been used as an effective method to investigate the organic non-linear optical (NLO) materials. Recent research works have illustrated that the organic non-linear optical materials are having high optical non-linearity than inorganic materials[28]. In the presence of an applied electric field, the energy of a system is a function of the electric field. Polarizabilities and hyperpolarizabilities characterize the response of a system in an applied electric field[29]. They determine not only the strength of molecular interactions but also the cross sections of different scattering and collision processes, as well as the NLO properties of the system[30]. For this subject, in this study the electronic dipole moment, molecular polarizability, anisotropy of polarizability and molecular first hyperpolarizability of present compound were investigated. The polarizability α and the hyperpolarizability β and the electric dipole moment μ of compound are calculated by finite field method using B3LYP/6-311++G (d, p) basis set available in Gaussian 03 package. The polarizability and hyperpolarizability tensors (α xx, α xy, α yy, α xz, α yz, α zz and β xxx, β xxy, β xyy, β yyy, β xxz, β xyz, β yyz, β xzz, β yzz, β zzz) can be obtained by a frequency job output file of Gaussian. However α and β values of Gaussian output are in atomic units (a.u.) so they have been converted into electronic units (esu) (α ; 1 a.u.=0.148 2× 10-24 esu, β ; 1 a.u.=8.639 3× 10-33 esu).
The calculated values of dipole moment (μ ), polarizability (α ) and first hyperpolarizability (β ) are tabulated in Table 2. The magnitude of the molecular hyperpolarizability β , is one of key factors in NLO system. The calculated first static hyperpolarizability β tot value is equal to 14.69× 10-30 esu. Domination of particular component indicates on a substantial delocalization of charges in that direction. It is noticed that in β xxx direction, the biggest values of hyperpolarizability are noticed and subsequently delocalization of electron cloud is more in that direction. The maximum β value may be due to p-electron cloud movement from donor to acceptor which makes the molecule highly polarized and the intramolecular charge transfer possible. The μ , α and β of 4CPNMPN are 1.501 5 D, 3.687× 10-23 esu and 1.469× 10-29 esu, respectively, obtained by B3LYP/6-31++G (d, p) method. Urea is the prototypical molecule used in the study of the NLO properties of the molecular systems. Therefore urea was used frequently as a threshold value for comparative purposes. In this study, the total dipole moment, polarizability and the calculated β value of the molecule is greater than urea (the μ , α and β of urea are 1.373 2 D, 3.831 2× 10-24 esu and 0.780 3× 10-30 esu). Hence the first order hyperpolarizability (β tot) of 4CPNMPN with B3LYP/6-31++G (d, p) basis set is very much greater than the urea value. From the computation the high values of 4CPNMPN are probably attributed to the charge transfer existing between the phenyl rings within the molecular skeleton. We conclude that the compound is an attractive object for future studies of non-linear optical properties.
3 Conclusion
Geometry of 4CPNMPN molecule was optimized using DFT at B3LYP level of theory employing 6-31++G(d, p) basis set. Attempts have been made in the present study for investigation of structure, molecular orbitals, corrosion inhibition studies and NBO analysis of the compound 4-chlorophenyl)-N-(4-methylphenyl) nitrone. Molecular orbital analysis shows that region of HOMO level spread over the chlorine aom and phenyl group, but it spreads over entire molecule for the LUMO. The Natural Bond Orbital (NBO) analysis provided the detailed insight into the type of hybridization and the nature of bonding in the molecule. The investigated efficiency of the compound has been investigated using DFT quantum chemical approach. From the results, it can be concluded that the compound can act as good corrosion inhibitor. The predicted first order hyperpolarizability shows that the molecule might have a reasonably good nonlinear optical (NLO) behaviour.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|