

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2-巯基-5-硝基苯并咪唑振动光谱的密度泛函理论研究
[陈玉锋1  , 邵长斌
, 邵长斌1 , 左明辉1 , 庄志萍1 , 赵冰2 ]
, 邵长斌|
|


作者简介: 陈玉锋, 1980年生, 牡丹江师范学院化学化工学院讲师 e-mail: chenyf11@mails.jlu.edu.cn
选用密度泛函理论中的B3LYP杂化泛函, 在B3LYP/6-31++g(d,p)(C, H, N, S)水平下, 优化了2-巯基-5-硝基苯并咪唑分子(MNBMZ)的结构, 优化结果表明, 2-巯基-5-硝基苯并咪唑分子是一个近平面结构。 通过频率计算, 获得了2-巯基-5-硝基苯并咪唑分子(MNBMZ)的拉曼光谱, 并和实验获得的拉曼光谱图进行了对比, 200~800 cm-1波数段实验获得的拉曼谱带波数和理论计算波数相比, 有一定程度的蓝移, 800~1 800 cm-1波数段实验获得的拉曼谱带波数和理论计算波数相比, 发生了一定的红移。 对实验和理论计算光谱主要振动峰进行线性回归拟合, 相关系数 r=0.998, 标准偏差14.98。 实验和理论计算获得的拉曼光谱图基本上是一致的, 表明本文选取的DFT理论计算方法是可靠的。 结合VEDA4软件对2-巯基-5-硝基苯并咪唑分子的拉曼谱带简正振动模式进行了指认。 此外, 分析并讨论了2-巯基-5-硝基苯并咪唑分子(MNBMZ)前线轨道及HOMO, LUMO轨道的组成, HOMO和LUMO轨道能级差为3.31 eV, 电子有从HOMO跃迁到LUMO的趋势。 HOMO轨道中S原子的贡献是52.53%, LUMO轨道中硝基N和O原子的贡献分别为23.03%, 19.97%和19.36%。 采用含时密度泛函理论(time dependent density functional theory, TDDFT)对2-巯基-5-硝基苯并咪唑分子(MNBMZ)的激发态进行了计算分析, 计算结果表明甲醇溶剂中2-巯基-5-硝基苯并咪唑分子(MNBMZ)理论计算的吸收波长为213, 281和437 nm; 实验获得的吸收波长223, 272和353 nm。 对研究2-巯基-5-硝基苯并咪唑分子的性质, 提供了理论基础。
Raman spectroscopy and optimized geometry of the 2-Mercapto-5-nitrobenzimidazole (MNBMZ) molecule had been calculated at density functional B3LYP level using 6-31++g(d,p) basis set in this paper. Raman spectrum was obtained from the calculation results of the frequencies, and compared with the experimental Raman spectrum. The compared results showed that there is a blue shift in the range of 200~800 cm-1, however in the range of 800~8 800 cm-1, there is a red shift. A line of best fit of the experimental Raman frequences versus the calculated ones in the range of 200~1 800 cm-1, the correlation coefficient and the standard deviation were 0.998 and 14.98. The vibrational mode was assigned on the basis of potential energy distribution (PED) through the VEDA4. In addition, the Frontier HOMO-LUMO orbital and the compositions were discussed based on the calculated results, and the HOMO-LUMO gap was estimated to be 3.31 eV, which indicated that the electron will transfer from the HOMO to LUMO. The contribution of S to the HOMO orbital was 52.53%, and the contribution of N and O in nitryl was 23.03, 19.97, 19.36 to the HOMO orbital. The excited states were calculated by TDDFT, and the results showed that the adsorption wavelength is 213, 281 and 437 nm, but it is 213, 272 and 353 nm especially from the experimental spectrum in the methyl alcohol. This study provided a theoretical support for the analysis of MNBMZ.
苯并咪唑是含两个氮原子的芳香杂环化合物, 具有咪唑环和苯环共平面的结构, 这种特殊的芳香体系使其易于与生物体内的受体和酶等形成氢键, 与金属离子配位以及发生π — π 相互作用等, 具有显著的生物活性, 因而具有重要的医用价值[1, 2]。 同时苯并咪唑类化合物还可以用作金属缓蚀剂、 荧光探针、 过渡金属配体、 有机合成反应的中间体等而应用广泛[5, 6, 7, 8, 9, 10]。
密度泛函理论(density functional theory, DFT)和实验数据相结合常被用来研究分子的几何构型和振动光谱[11, 12, 13], B3LYP方法因为具有较高的精确度和对计算资源要求不高, 应用较广泛。 本文采用B3LYP杂化泛函, 6-31++g(d, p)基组[14, 15], 计算了2-巯基-5-硝基苯并咪唑的拉曼光谱, 和实验获得的拉曼光谱图进行了对比, 利用VEDA4软件对2-巯基-5-硝基苯并咪唑的简正振动模式进行了指认[16]。 并结合TDDFT计算分析研究了MNBMZ分子的吸收光谱和激发态。
理论计算采用Gaussian09量子化学程序包[17], 分子构型用Gauss View5.0构造, 利用Gaussian09程序包, B3LYP/6-31++g(d, p)水平上对2-巯基-5-硝基苯并咪唑分子的几何结构进行优化和频率计算。 简正振动模式分析采用VEDA4软件完成。
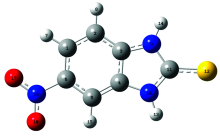
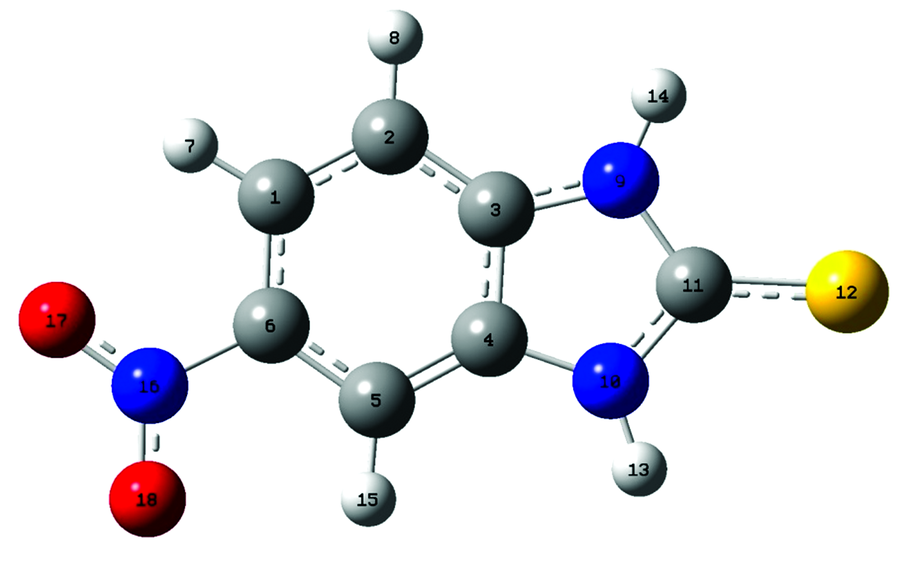 | 图1 2-巯基-5-硝基苯并咪唑分子结构模型示意图Fig.1 Optimized structure of MNBMZ |
2-巯基-5-硝基苯并咪唑分子的常规拉曼光谱(NRS)采集使用B& W Tek公司的BWS415-785S型i-Raman便携拉曼光谱仪, TE制冷线性CCD阵列检测器, 激发光源波长785 nm, 积分时间5 s, 积分1次。 所有数据处理采用软件NGSLabSpec1-Origin中的Baseline correction 进行处理, 并用Origin 7.5工具作图; 紫外光谱采集使用上海元析仪器有限公司的UV-5500PC紫外-可见分光光度计。 为了和实验获得的拉曼光谱对比, 计算得到的MNBMZ分子的拉曼光谱图的拉曼强度扩大了一定的倍数。
优化结果中没有发现虚频, 表明优化得到的结构是2-巯基-5-硝基苯并咪唑分子的稳定构型。 计算结果表明, C1H6N16O17, C5C6N16O18, H14N9C11S12, H13N10C11S12的二面角均接近0.00° 或180.00° , 表明2-巯基-5-硝基苯并咪唑分子结构是近平面结构。
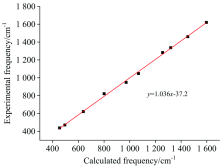
MNBMZ分子共有48个振动模式, 17个伸缩振动模, 18个弯曲振动模, 15个扭转振动模。 利用VEDA4软件对2-巯基-5-硝基苯并咪唑的简正振动模式进行了指认, 结果见表1。 本文选取MNBMZ分子的200~1 800 cm-1的实验和理论计算数据作拉曼光谱图见图2, 200~800 cm-1波数段实验获得的拉曼谱带波数和理论计算波数相比, 有一定程度的蓝移, 800~1 800 cm-1波数段实验获得的拉曼谱带波数和理论计算波数相比, 发生了一定的红移, 实验和理论计算获得的拉曼光谱图基本上是一致的。 对实验和理论计算光谱主要振动峰进行线性回归拟合, 见图3, 结果表明, 相关系数r=0.998, 标准偏差14.98, 两者吻合较好, 表明本文选取的DFT理论计算方法及水平是可靠的。
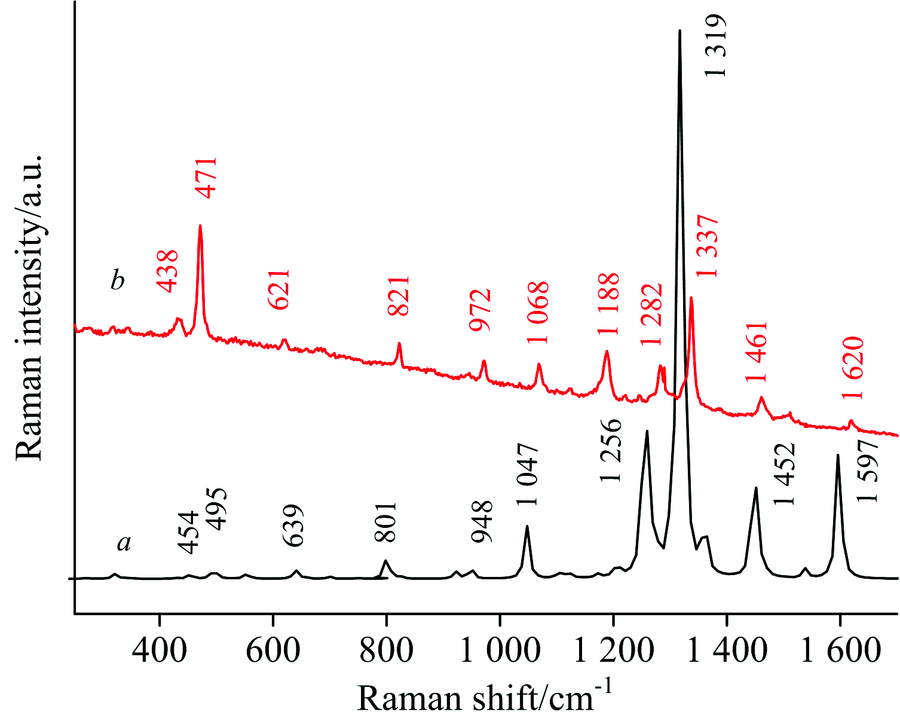 | 图2 MNBMZ理论计算和实验的拉曼光谱图Fig.2 The Theoretical (a) and Experimental (b) Raman spectrum of MNBMZ |
| 表1 MNBMZ的理论频率、 实验频率振动归属 Table 1 Theoretical, Experimental vibrational frequencies and assignment of MNBMZ |
 | 图3 MNBMZ分子的主要实验和理论计算频率线性回归拟合Fig.3 The linear regression fitting between main experimental and Theoretical Raman frequencies of MNBMZ |
438 cm-1归属于苯环上C— C— C的面内不对称弯曲振动; 471 cm-1是拉曼谱图中的较强峰, 归属于C— S伸缩, C— H, N— H的面内弯曲振动; 821 cm-1归属于C— C伸缩和苯环上C— C— C的面内不对称弯曲振动; 972 cm-1被指认为C— N伸缩以及N— C— N的面内弯曲振动; 1 068 cm-1被指认为C— C伸缩、 C— N伸缩以及苯环上C— H的面内弯曲; 1 282 cm-1峰被指认归属于的C— C伸缩C— N伸缩和H— C— C, H— C— N的面内弯曲振动; 1 337 cm-1是拉曼谱图中最强的峰, 归属于C— C伸缩、 C— N伸缩、 C— S伸缩和C— N— C的面内弯曲振动、 C— C— C的面内弯曲振动; 1 461 cm-1归属于C— C伸缩、 C— N伸缩和C— H、 N— H的面内弯曲振动; 1 620 cm-1的拉曼峰被指认为C— C伸缩振动、 C— H面内弯曲振动。
分子中的前线轨道即最高占据轨道(highest occupied molecular orbital, HOMO)和最低空轨道(lowest unoccupied molecular orbital, LUMO)可以提供分子的电子吸收光谱性质、 化学亲电亲核性能等重要的分子信息。 MNBMZ分子的前线轨道见图4。 HOMO和LUMO轨道能级分别为-6.37和-3.06 eV, 能级差为3.31 eV, 电子有从HOMO跃迁到LUMO的趋势。 HOMO轨道中S原子的贡献是52.53%, LUMO轨道中硝基N和O原子的贡献分别为23.03%, 19.97%和19.36%[19]。
 | 图4 MNBMZ分子的前线轨道Fig.4 The frontier orbital of MNBMZ |
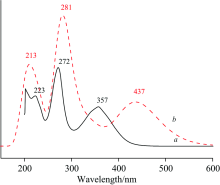
2-巯基-5-硝基苯并咪唑分子(MNBMZ)的TDDFT激发态计算所得到的吸收光谱见图5。 TDDFT激发态计算所得到的2-巯基-5-硝基苯并咪唑分子(MNBMZ)的结构轨道跃迁、 垂直激发能、 波长和谐振强度结果见表5。 由图5可以看出, 甲醇溶剂中2-巯基-5-硝基苯并咪唑分子(MNBMZ)理论计算的吸收波长为213, 281和437 nm; 实验获得的吸收波长223, 272和353 nm。
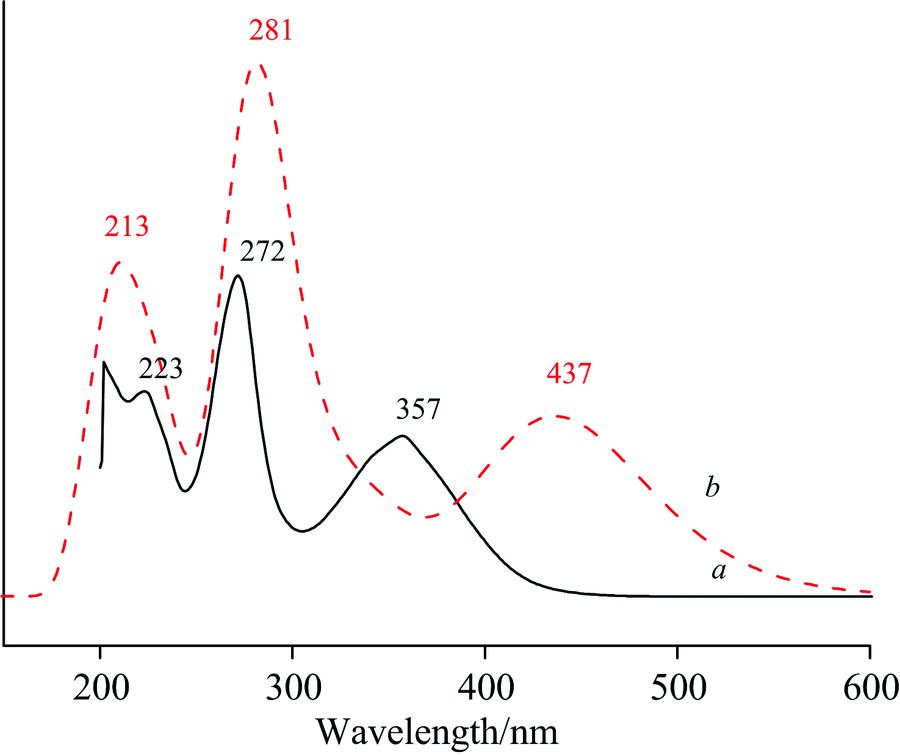 | 图5 MNBMZ分子的紫外-可见吸收光谱a: 实验; b: 理论Fig.5 The simulated adsorption spectra of MNBMZa: Experimental; b: Theoretical complex |
| 表5 MNBMZ的激发态 Table 5 The several singlet excited states of MNBMZ |
选用密度泛函理论中的B3LYP杂化泛函, 在B3LYP/6-31++g(d, p)(C, H, N, S)水平下, 优化了2-巯基-5-硝基苯并咪唑分子(MNBMZ)的结构, 通过频率计算, 获得了2-巯基-5-硝基苯并咪唑分子(MNBMZ)的拉曼光谱, 实验和理论计算获得的拉曼光谱图基本上是一致的, 对实验和理论计算光谱主要振动峰进行线性回归拟合, 相关系数r=0.998, 标准偏差14.98, 两者吻合较好, 表明本文选取的DFT理论计算方法是可靠的, 并结合VEDA4软件对2-巯基-5-硝基苯并咪唑分子的简正振动模式进行了指认。 此外, 分析并讨论了2-巯基-5-硝基苯并咪唑分子(MNBMZ)前线分子轨道及HOMO, LUMO轨道的原子组成, 分析了Hirsheld电荷, 通过TDDFT计算了吸收光谱和激发态。 对研究2-巯基-5-硝基苯并咪唑分子的性质, 提供了理论基础。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|