{kind=link}
{kind=link}
电感耦合等离子体串联质谱法分析麻疯树油中的多元素
[江波1, 3  , 黄建华
, 黄建华2, * ]
, 黄建华]
|
|


作者简介: 江 波, 1974年生, 长江师范学院生命科学与技术学院讲师 e-mail: jblmz74@126.com
工业化和现代化进程的加快消耗了大量能源, 对能源的高度依赖性导致了全球化石能源需求的快速增长, 随着非再生化石能源的日渐枯竭, 迫切需要大力发展可再生能源以调整现有能源结构。 作为国际上研究最多的生物柴油, 麻疯树油是国内外公认的最有可能替代化石能源的再生能源, 具有极大的开发潜力。 麻疯树油中的微量元素在燃烧过程中会影响发动机的性能, 并在尾气排放过程中决定了对环境所造成的污染程度。 本文以获得麻疯树油中多元素的含量为目的, 建立应用电感耦合等离子体串联质谱(ICP-MS/MS)法准确测定麻疯树油中低水平Na, Si, P, S, Cl, K, Ti, V, As含量的分析方法。 采用微波密闭消解系统, 依次向麻疯树油样品中加入硝酸和双氧水进行消解。 详细地研究了各待测元素在不同分析模式下检出限(DL)和背景等效浓度(BEC)的变化情况, 在MS/MS模式下, 通过向碰撞/反应池(CRC)中加入反应气可以完全消除质谱干扰。 选择O2为反应气, P+, S+, Ti+, V+, As+与O2的反应均为放热过程, 能发生质量转移自发生成PO+, SO+, TiO+, VO+, AsO+, 利用O2质量转移法消除质谱干扰; 选择H2为反应气, Cl+与H2反应能自发生成Cl$H^{+}_{2}$, 利用H2质量转移法消除质谱干扰, 而Na+, Si+, K+均不能与H2发生质量转移反应, 利用H2原位质量法消除质谱干扰。 选择Sc为内标元素校正了分析过程中的基体效应。 通过考察不同反应气流速下各元素的BEC变化, 优化了反应气流速, O2的最佳流速为0.45 mL·min-1, H2的最佳流速为7.5 mL·min-1。 在优化的实验条件下获得Na, Si, P, S, Cl, K, Ti, V, As的检出限分别为6.41, 37.3, 24.6, 118, 530, 7.96, 7.61, 0.34, 3.20 ng·L-1, 各元素在0~50 μg·L-1范围内的线性相关系数( R2)≥0.999 8, 方法具有良好的线性关系。 采用三水平加标回收实验来验证方法的准确性和精密度, 所有元素的加标回收率在91.2%~108%之间, 相对标准偏差(RSD)为1.9%~4.6%, 表明所建立的方法准确性好, 精密度高。 通过对来自中国不同地区的4个麻疯树油样品进行测定, 结果显示, 4个麻疯树油样品中P含量≤164 ng·g-1, S含量≤2310 ng·g-1, 碱(Na+K)含量≤1 690 ng·g-1, 三项指标均达到了中国生物柴油调和燃料国家标准, 欧Ⅳ生物柴油标准, 德国生物柴油标准和美国生物柴油标准。 这项研究为麻疯树油中多种微量元素的准确分析提供了一种方便可行的新方法, 为麻疯树油的质量控制和安全应用提供了科学的理论依据。
According to the development of industrialization and modernization, much more conventional fossil energy resource is excess consumed, with the depletion of non-renewable energy resource, the development of new renewable energy was urgently needed. As one of the hottest concerned biodiesel resources, Jatropha curcas L. oil was thought as the most possible renewable energy to replace conventional fossil energy resource. The trace elements in Jatropha oil might affect the performance of engine, and deteriorate environment. In order to obtain the contents of multi-elements in Jatropha curcas L. oil, an analytical method was established for accurately determination of Si, P, S, Cr and As in Jatropha curcas L. oil by using inductively coupled plasma tandem mass spectrometry (ICP-MS/MS). Jatropha oil was processed by using microwave-assisted acid-digested reaction with nitric acid and hydrogen peroxide. The changes of detection limit (DL) and background equivalent concentration (BEC) for multi-elements in different analysis modes were optimized in detail. In the MS/MS mode, O2 was introduced into the collision reaction cell (CRC), then Si+, P+, S+, Cr+ and As+ were reacted with O2 to generate Si$O^{+}_{2}$, PO+, SO+, CrO+ and AsO+ respectively; thus mass spectrometry interference can be eliminated by mass shift caused by reactions. As Cl+ can reaction with H2 to form Cl$H^{+}_{2}$, while Na+, Si+, K+ elements cannot reaction with it, the mass shift caused by reaction with H2 was selected to eliminate mass spectrum interference. Sc was selected as internal standard element to correct the matrix effects. The flow rates in the CRC of different reaction gas were optimized by considering the background equivalent concentration (BEC) of analytes, and the best gas flow rate for O2 and H2 were 0.45 mL·min-1, and 7.5 mL·min-1, respectively. The DL for Na, Si, P, S, Cl, K, Ti, V, and As were 6.41, 37.3, 24.6, 118, 530, 7.96, 7.61, 0.34, and 3.20 ng·L-1, respectively, under the optimized conditions. The linear correlation coefficient ( R2) of analytes were ≥0.999 8 in the range 0~50 μg·L-1. The recovery of all elements ranged from 91.2% to 108%, and the relative standard deviation (RSD) was ranged from 1.9% to 4.6%. These results showed that the proposed method was accuracy and precise. Analytical results obtained from different original Jatropha curcas L. oil showed that the contents of element P, S, and (Na+K) were ≤164, 2 310, and 1 690 ng·g-1, respectively. These indexes were much lower than those of the contents in the conventional fossil energy resource, and reached the standards of Chinese national standard of biodiesel, European IV biodiesel standard, German biodiesel standard, and American biodiesel standard. This study proposed a new approach for the determination of multi-element in Jatropha curcas L. oil with convenience and feasibility, and provided a scie.pngic basis for the quality control and safety application of Jatropha curcas L. oil.
麻疯树为大戟科小乔木或灌木, 是国际上公认能生产生物柴油的能源植物[1], 也是我国最有种植潜力的绿色能源树种, 其果实含油量高达60%, 所提炼出来的麻疯树油与石化柴油成分相近, 在闪点, 凝固点, CO排放量, 颗粒值等关键指标上均优于国内零号柴油, 达到欧Ⅳ 生物柴油的标准, 具有润滑功能强, 安全性能高, 燃料性能佳以及低温启动性能好等优点[2, 3, 4]。 麻疯树油中的微量元素主要来源于果实原料, 另外在在麻疯树油的提炼, 精制过程中, 由于催化剂及其载体使用有带入微量元素的可能[5, 6, 7], 鉴于麻疯树油中的微量元素在燃烧过程中会沉积在发动机缸体, 活塞及汽缸壁上, 造成堵塞和磨损从而缩短发动机使用寿命, 同时决定了排放尾气中微量元素的浓度对环境的影响程度。 因此, 有必要对麻疯树油中微量元素的含量进行检测, 建立快速准确测定麻疯树油中微量元素含量的分析方法具有十分重要的应用价值。
目前, 有关生物柴油中微量元素的分析方法主要包括原子吸收光谱(AAS)法[8], 电感耦合等离子体发射光谱(ICP-OES)法[9]和电感耦合等离子体质谱(ICP-MS)法[10], 在这些分析方法中, ICP-MS法的灵敏度, 检出限以及消除干扰的能力均优于AAS法和ICP-OES法, 并能提供可靠的同位素分析信息, 能满足生物柴油中绝大多数微量元素的分析要求, 但ICP-MS所面临的质谱干扰仍然限制了部分元素的准确测定, 在减少或消除质谱干扰的技术创新过程中, 开发出多项重要的技术[11, 12, 13], 其中碰撞/反应池(CRC)技术提供了消除质谱干扰的通用方法, 在碰撞模式下, 可以通过设置偏置电压借助动能歧视消除干扰, 在反应模式下, 可以通过反应化学上的差异消除干扰, 随着电感耦合等离子体串联质谱(ICP-MS/MS)的推出, 采用CRC技术消除干扰的能力得到了显著提升[14, 15, 16]。 ICP-MS/MS法是在等离子体(ICP)和CRC间增设质量过滤器(Q1), 与CRC和检测器间的质量过滤器(Q2)构成了三重串联四极杆, 通过对ICP和CRC提取的离子进行选择, Q1只允许指定质荷比(m/z)的离子进入CRC, 而将其他m/z的离子排除在外, Q2只选择来自CRC的目标m/z离子, 阻挡了其他m/z的离子, 从而精准控制CRC中的离子或分子发生选择性化学反应[17, 18, 19]。 本文应用微波消解对麻疯树油进行预处理, 利用ICP-MS/MS法测定了其中的Na, Si, P, S, Cl, K, Ti, V, As等9个微量元素, 旨在为麻疯树油中微量元素的快速准确分析提供一种新方法。
1 000 mg· L-1的Na, Si, P, S, Cl, K, Ti, V, As单元素标准溶液, 国家标准物质中心; 10 mg· L-1的Sc内标溶液, 国家标准物质中心; 1 μ g· L-1的Li, Y, Ce, Tl调谐溶液, 美国Agilent公司; 硝酸和双氧水均为优级纯, 北京化学试剂研究所; 高纯氧气, 长沙方罡气体有限公司; Milli超纯水。
8800型ICP-MS/MS仪: 美国Agilent公司; 通过调谐优化后ICP-MS/MS的工作条件为: 射频(RF)功率1 550 W, 等离子模式, 低基质; 等离子气流速, 15.0 L· min-1; 载气流速, 0.7 L· min-1; 补偿气流速, 0.2 L· min-1; 采样深度, 8.0 mm; 质谱模式, MS/MS; CRC气体, O2, H2; O2流速, 0.40 mL· min-1; H2流速, 7.5 mL· min-1; 八极杆偏置电压, -14 V(O2), -10 V(H2); KED偏置电压, -5 V(O2), 0 V(H2原位质量), -5 V(H2质量转移); 质谱分析总时间, 120 s(包括不同反应气之间进行切换的平衡时间30 s)。 MARs 5微波消解仪: 美国CEM公司。
称取约0.5 g麻疯树油样品于微波消解罐中, 依次加入4 mL 硝酸和1 mL双氧水, 敞开罐盖于180 ℃预消解20 min后置于微波消解仪的腔体内进行消解。 微波消解程序为: 功率1 600 W, 3 min内升温至100 ℃后保持3 min; 7 min内升温至150 ℃后保持3 min; 5 min内升温至170 ℃后保持3 min; 5 min内升温至190 ℃后保持10 min。 消解完毕后自动冷却至室温, 用超纯水转移至100 mL容量瓶中定容, 采用相同的过程制备空白溶液。
采用Na, Si, P, S, Cl, K, Ti, V, As单元素标准溶液以4%(V/V)的硝酸为介质, 配制系列标准混合溶液, 在优化的质谱工作条件下分别对样品溶液, 标准溶液和空白溶液进行测定, 以待测元素与内标元素的信号强度的比值对各元素对应的浓度建立标准曲线, 根据标准曲线计算出样品溶液中各元素的浓度。 在测定过程中, 所有上机测试溶液(样品溶液, 标准溶液和空白溶液)均通过标准内标 “ T” 型混合接头在线加入1 mg· L-1的Sc内标元素。
在单四极杆(SQ, Q1无过滤功能, 相当于传统的ICP-QMS)模式下分别采用无气和He碰撞方式, 在MS/MS(Q1和Q2均具有过滤功能)模式下分别采用He碰撞, O2反应和H2反应方式, 考察了5种不同质谱分析模式下9个待测元素的检出限(DL)和背景等效浓度(BEC)的变化情况, 其中DL为连续测定11次空白溶液[4%(V/V)的硝酸]的3倍标准偏差所对应的浓度, BEC为空白溶液的平均测定结果所对应的浓度[20], 结果见表1。
| 表1 不同分析模式下各元素的DL和BEC(ng· L-1) Table 1 Instrument detection limits (DLs) and background equivalent concentrations (BECs) for analytes determined using different analysis mode (ng· L-1) |
从表1可以看出, 在SQ模式下, 采用He为碰撞气, 借助动能歧视(KED)作用可以消除多原子离子干扰, 与无气方式相比较, 元素Na, P, K, Ti, V, As的BEC和DL均变小, 其中Ti和V效果显著, 而对于非金属难电离元素Si, S, Cl, 面临严重而复杂的质谱干扰, 采用无气方式和He碰撞方式均无法进行测定, 即使在MS/MS模式下采用H2为反应气, 仍然无法实现S的测定; 对比不同模式下的He碰撞方式, 在 MS/MS模式下Na, P, K, Ti, V, As的BEC和DL均小于SQ模式, 说明在MS/MS模式下, 通过Q1消除了大量干扰离子, 质谱通道更加畅通, 待测元素的传输效率高, 能量损失小。
在MS/MS模式下, 所有元素均可采用反应方式消除干扰。 元素S只能使用O2为反应气进行测定, 而对于元素P, Ti, V, As, 采用O2为反应气时, 各元素的BEC和DL均小于H2反应模式, 因此, 实验选择O2反应模式消除元素P, S, Ti, V, As分析过程中的质谱干扰; 采用H2为反应气, 元素Na, Si, K的BEC和DL均小于O2反应模式, 元素Cl的BEC虽然略大于O2反应模式, 但DL远小于O2反应模式, 实验最终确定元素Na, Si, Cl, K的测定采用H2反应模式进行分析(见图1)。
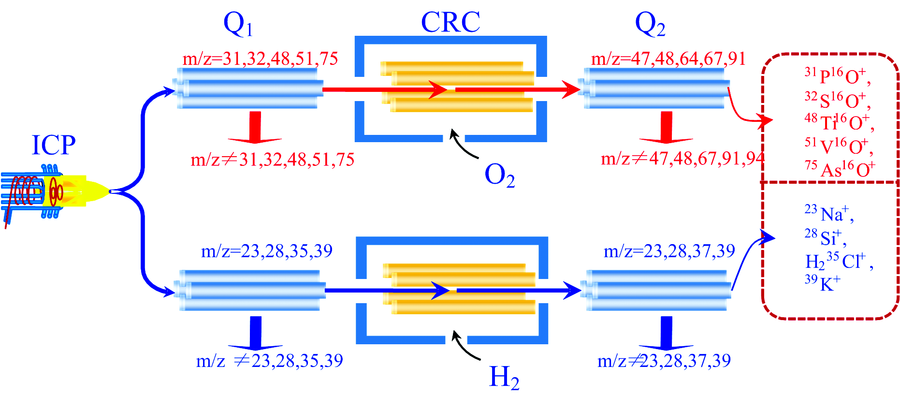 | 图1 ICP-MS/MS在MS/MS模式下的质量转移原理Fig.1 Mechanism of mass-shift in the MS/MS mode by ICP-MS/MS |
使用O2质量转移法测定麻疯树油中的31P, 所面临的主要质谱干扰为14N16OH, 15N16O, 15N15NH, 12C18OH, 14N17O, 31P与O2发生质量转移反应的动力学方程为: 31P++O2→ 31P16O++O(Δ Hr=-3.17 eV), 放热反应数据表明, 31P+能与O2自发反应高效地转移为31P16O+, 而31P的主要干扰离子均不能与O2发生反应, 因此可以通过质量转移改变待测元素的m/z消除干扰, 设置Q1的m/z=31, 将m/z≠ 31的所有干扰离子排出质谱通道, 设置Q2的m/z=47, 则31P+与O2反应的产物离子31P16O+能顺利通过Q2进入质谱检测器, 而干扰离子14N16OH+, 15N16O+, 15N15NH+, 12C18OH+, 14N17O+仍然维持原有的m/z=31, 通过Q2过滤被阻止进入质谱检测器, 从而消除干扰。 与31P+相似, 32S+, 48Ti+, 51V+, 75As+与O2发生质量转移反应的动力学方程均为放热反应, 而这些元素的干扰离子也不与O2发生反应, 设置质量对Q2=Q1+16, 利用O2质量转移反应, 分别生成相应的氧化物消除干扰。
28Si所面临的质谱干扰主要来自N, C, O所形成的14N2和12C16O, 采用H2为反应气, 28Si与H2反应的动力学方程为: 28Si++H2→ 28SiH++H(Δ Hr=1.30 eV), 吸热反应数据表明, 28Si+不与H2发生反应, 而干扰离子与H2反应的动力学方程为: 14
| 表2 在MS/MS模式下待测元素质谱干扰的消除方法 Table 2 Elimination method of interference of analyte in the MS/MS mode |
反应气流速决定CRC中反应产物浓度, 影响干扰的消除程度, 优化反应气流速可以将各待测元素的BEC降低至较低水平。 考察了不同O2流速在4%(V/V)的硝酸溶液中P, S, Ti, V, As的BEC变化情况, 当O2流速分别达到0.34, 0.40, 0.38, 0.42, 0.40 mL· min-1时, 对应元素P, S, Ti, V, As的BEC具有最小值, 本实验中采用0.40 mL· min-1的最佳O2流速; 考察了不同H2流速在4%(V/V)的硝酸溶液中Na, Si, Cl, K的BEC变化情况, 当这些元素的BEC处于最小值时所对应的H2流速分别为6.5, 7.5, 7.0, 6.5 mL· min-1, 基于Cl的测定过程中利用了H2二次质量转移, 而Cl的电离能高达12.97 eV, 在等离子体中电离度极差, 信号强度极低, 为确保H2二次质量转移过程中尽可能生成更多的35Cl
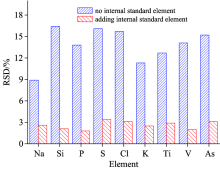
麻疯树油经微波消解后得到的样品溶液, 在物理化学性质上与校准标准溶液存在较大差异, 分析过程中表现为溶液提升, 样品传输以及雾化效率存在差异导致误差, 实验选择内标元素Sc做参照物校正这些误差。 在MS/MS模式下, 反应气及反应方式的不同, 选择内标元素的产物离子也不同, 采O2为反应气, 内标元素Sc与O2的动力学方程为: 45Sc++O2→ 45Sc16O++O(Δ Hr< 0), 因此, 利用O2质量转移法进行分析的P, S, Ti, V, As, 所选择内标离子为45Sc16O+; 对于采用H2质量转移法消除干扰的Cl, 选择45Sc16H+为内标离子进行测定, 而对于采用H2原位质量法进行测定的Na, Si, K, 则选择45Sc+为内标离子。 图2为麻疯树油加入内标元素前后各待测元素相对标准偏差(RSD, n=11)的变化情况, 从图2可以看出, 内标元素的加入显著改善了各待测元素的RSD, 基体效应得到了校正。
 | 图2 内标元素对基体效应的校正效果Fig.2 Correction effect of internal standard elements on matrix effect |
将浓度分别为0.0, 0.2, 1.0, 5.0, 20 μ g· L-1的系列标准溶液进行测定, 各待测元素在0~20 μ g· L-1的线性范围内的相关系数(R2)≥ 0.999 8, 验证了所建立的分析方法具有良好的线性关系。 Na, Si, P, S, Cl, K, Ti, V, As的检出限分别为6.41, 37.3, 24.6, 118, 530, 7.96, 7.61, 0.34, 3.20 ng· L-1。 为验证方法的准确性, 考察了各元素的加标回收率, 选取麻疯树油样品进行加标回收实验, 分别在样品中加入200, 1 000, 5 000 ng· L-1三水平的标准溶液, 经微波消解处理后测定, 每个样品连续测定11次, 结果见表3。 所有元素的低, 中, 高三水平的加标回收率在91.2%~108%之间, RSD为1.9%~4.6%, 验证了方法的准确性和精密度均能满足分析要求。
| 表3 应用ICP-MS/MS测定麻疯树油中多元素的加标回收实验 Table 3 Mean results for spike recovery experiments in Jatropha curcas L. oil for determination multi-element of using ICP-MS/MS |
采用本工作建立的ICP-MS/MS法对来自不同地区的4个麻疯树油(样品编号A, B, C, D)连续测定10次, 分析结果见表4。 4个麻疯树油样品中Na含量为590~1 640 ng· g-1, Si含量为98.7~275 ng· g-1, P含量为40.3~164 ng· g-1, S含量为870~2 310 ng· g-1, Cl含量为20.1~53.9 ng· g-1, K含量为51.6~330 ng· g-1, Ti含量为2.73~6.14 ng· g-1, V含量为0.36~0.91 ng· g-1, As含量为3.05~11.8 ng· g-1, 碱(Na+K)含量为864~1 690 ng· g-1。 4个麻疯树油中P, S, 碱(Na+K)三项指标均达到了我国生物柴油调和燃料国家标准, 欧Ⅳ 生物柴油标准, 德国生物柴油标准和美国生物柴油标准, 而麻疯树油中的Si, Cl, Ti, V, As的含量也处于很低水平。
| 表4 麻疯树油样品的分析结果(ng· g-1, 平均值正负偏差, n=11) Table 4 Analysis results obtained for Jatropha curcas L. oil samples (ng· g-1, mean± standard deviation, n=11) |
建立了利用ICP-MS/MS准确测定麻疯树油中多元素的新方法。 在MS/MS模式下, 分别使用O2和H2为反应气, 利用质量转移反应能有效消除质谱干扰, 可测定麻疯树油中的九种元素。 对于P, S, Ti, V, As的测定, 采用O2反应模式消除干扰非常有效; 对于Na, Si, Cl, K的测定, 采用H2反应模式能完全消除干扰。 在本研究所分析的4个麻疯树油样品中, P, S, 碱(Na+K)三项指标均达到了中国、 欧盟、 德国和美国生物柴油标准, 而Si, Cl, Ti, V, As的含量也处于很低水平。 方法具有分析速度快, 准确性好, 精密度高的特点, 适合于大批量样品的分析, 能为麻疯树油品质的评价和安全应用提供科学的理论依据。
致谢: 本工作得到了湘产大宗药材品质评价湖南省重点实验室, 湖湘中药资源保护与协同创新中心, 湖南省药食同源功能性食品工程技术研究中心和湖南省中药不良成分快速检测及脱除工程技术研究中心的资助和支持。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|