


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
氧化槐果碱结构的测定及其振动圆二色谱研究
[孙宁杰1  , 蔡晓丽
, 蔡晓丽1 , 张越非1 , 池汝安1, * , YunjieXu2, * ]
, 蔡晓丽, YunjieXu]
|
|


作者简介: 孙宁杰, 1991年生, 武汉工程大学绿色化工过程省部共建教育部重点实验室硕士研究生 e-mail: ningjiesun@126.com
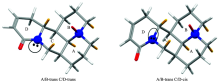
采用傅里叶红外光谱仪(FTIR)和振动圆二色谱仪(VCD)测量氧化槐果碱固体的红外光谱和振动圆二色谱。 采用密度泛函方法(DFT)分别在B3LYP/cc-PVTZ和B3LYP/6-311+G(d, p)水平下, 对气相中氧化槐果碱分子结构进行了优化, 然后在相同水平下计算了氧化槐果碱的红外光谱(IR)以及振动圆二色谱(VCD)。 将实验谱图与理论计算谱图进行对照, 以确定氧化槐果碱的实际构象以及构象分布。 由对比结果可知: 采用B3LYP/6-311+G(d,p)水平下, 以吉布斯自由能进行玻尔兹曼加权平均后的理论IR和VCD谱图与实际光谱图最为接近, 说明在常温下氧化槐果碱具有A/B-trans C/D-trans和A/B-trans C/D-cis两种优势构象, 且玻尔兹曼分布显示两种构象均以一定比例共存, 且通过对氧化槐果碱的两种优势构象分析, 发现A/B环的立体结构为: 椅式-椅式, C/D环的立体结构为: 椅式-沙发式。
Sophocarpidin was measured in thin film using a Fourier Transform infrared spectrometer (FTIR) with a vibrational circular dichroism (VCD) module, meanwhile, the theoretical IR and VCD spectroscopy were calculated at the B3LYP/cc-PVTZ and B3LYP/6-311+G(d,p) levels of theory in density functional theory (DFT) within the gas phase. The VCD spectroscopy can exactly obtain the conformational distribution of Sophocarpidin at room temperature and distinctly capture the information of each conformation for the sensitivity to stereoscopic structure of conformation within VCD spectra. It is essential to the drug analysis of sophocarpidin in the future. The conformational contribution at room temperature and structures were determined through the comparison of experimental and simulated spectra. The Boltzmann-weighted IR and VCD spectra based on free energy at the B3LYP/6-311+G(d,p) level of theory show good agreement with the observed experimental spectra. Two main conformations, i.e. A/B-trans C/D-trans and A/B-trans C/D-cis are ide.pngied. The ration of two conformations is 60% to 40%. The structure of A/B-ring and C/D-ring are chair-chair and chair-sofa respectively in both two dominant conformation of sophocarpidin.
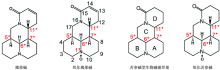
氧化槐果碱为槐果碱的氧化产物, 属于苦参碱型生物碱。 氧化槐果碱可从豆根槐属植物苦参或豆科植物广豆根提取而来。 氧化槐果碱具有重要的医药效用, 可用于治疗急性菌痢、 心律失常和淋巴结炎等疾病[1]。 Kuchkarov[2]、 Ueno[3]等先后从苦豆子、 苦参等植物中分离出氧化槐果碱。 目前, 氧化槐果碱的研究主要集中于分离、 提取、 纯化以及药理学等领域, 而对氧化槐果碱的绝对构象研究鲜为报道。 氧化槐果碱具有苦参碱型生物碱炭骨架, 苦参碱型生物碱分子骨架可看做两个喹诺里西啶环, 即A/B环和C/D环的稠合体。 图1为苦参型生物碱碳骨架, 以及氧化槐果碱和槐果碱的化学结构。 苦参型生物碱碳骨架具有四个碳原子手性中心C-5, C-6, C-7和C-11以及两个氮原子手性中心N-1和N-16。 所以苦参型生物碱碳骨架理论上有24× 2× 2(64)个立体异构体, 且可能存在四种构象, 即A/B-trans C/D-trans, A/B-trans C/D-cis, A/B-cis C/D-trans和A/B-cis C/D-cis, A/B环trans(cis)表示桥头氮原子上的N— O键方向和碳原子的直键氢方向相反(相同); C/D环trans(cis)表示桥头氮原子N-16的孤对电子方向和碳原子的直键氢方向相反(相同)[4](见图2)。 在1978年和1983年关于苦参碱型天然产物的报告中, Morinaga[5]和Sadykov[6]先后提出A/B环、 C/D环均以trans方式连接。 2000年, Rahman[7]等运用X射线单晶衍射法对12β -hydroxysophocarpine的研究中, 提出A/B环以trans方式连接, C/D环以cis方式连接。 2006年, Galasso[8]等在苦参碱型生物碱的计算研究中预测槐果碱会以C/D-trans和C/D-cis构象以一定比例同时存在于气相或液相中; 2016年, 蔡晓丽[4]等运用振动圆二色谱和量子化学计算确定槐果碱的绝对构型和构象分布: 在不同溶液状态下, A/B-trans C/D-trans和A/B-trans C/D-cis两种构象以一定比例共存; 同年, 张越非等在苦参碱型生物碱(苦参碱、 槐定碱以及氧化苦参碱)的研究中指出氧化苦参碱的构型(氧化苦参碱的化学结构如图1所示)为SSRR(分别对应C5, C6, C7和C11), 且通过计算精确地预测氧化苦参碱(苦参碱)两种构象在常温下玻尔兹曼分布的比例, 说明A/B-trans(A/B-cis)中N-1手性为S(R), C/D-trans(C/D-cis)中N-16手性为R(S)[9]。 相比于苦参型生物碱碳骨架, 氧化槐果碱是由喹诺里西啶A/B环的氮氧化合物和喹诺里西啶酮C/D环结合而形成, 其几何结构与槐果碱以及氧化苦参碱极其相似, 如图1所示。 所以, 可推测氧化槐果碱有四种可能构象(A/B-trans C/D-trans, A/B-trans C/D-cis, A/B-cis C/D-trans和A/B-cis C/D-cis)。 相对于槐果碱和氧化苦参碱的研究成果, 对氧化槐果碱的优势构象以及相关的玻尔兹曼分布尚未研究。 这个研究对未来药品的质量分析很有必要。
绝对构象(立体化学)的主要研究方法有基于手性试剂化学反应法、 NMR的Mosher法[10, 11]和X射线单晶衍射[12]。 相比于需要经过复杂的手性合成的有机合成法、 需要昂贵手性试剂且对测试分子有特殊要求的Mosher法以及需要高质量单晶的X射线衍射法, 基于手性分子的性质使其对平面偏振光的左、 右旋圆偏振光折射率以及吸收系数不同而获得手性分子结构信息的光谱学方法更可靠。 且由于光谱学方法不需要对目标化合物进行繁琐的合成反应、 不需要手性试剂的选择、 也不需要关注样品结晶与否而得到了广泛的运用。 旋光以及圆二色谱是光谱学法应用最广泛的两种方法。 圆二色谱(CD)经由紫外吸收区向红外吸收区的发展可分为电子圆二色谱(ECD)和振动圆二色谱(VCD), 其中旋光和电子圆二色谱均由于电子光学活性而引起, 然而振动圆二色谱则由振动光学活性作用而形成的光谱, 所以振动转动能级的跃迁使得振动圆二色谱更可靠, 且由于完整的手性分子的结构信息需通过实验光谱和量子化学理论光谱的对比而确认。 在理论计算层次, 振动圆二色谱仅包含基态电子能级的计算, 而电子圆二色谱的理论计算还包含了激发态电子能级的计算, 这意味着振动圆二色谱在理论计算光谱上也比电子圆二色谱更准确可靠[13], 故选择振动圆二色谱法为检测分子手性的方法。 近二十几年, 振动圆二色谱无论在实验技术还是理论计算都有了重大发展, 振动圆二色谱已经可以成熟地运用于分子结构的分析。 近几年, 振动圆二色谱结合量子化学计算的方法得到了广泛运用, 很多课题组[9, 14, 15]运用振动圆二色谱对分子结构和溶液中的非共价键的[16, 17]相互作用进行分析。 本研究联合运用傅里叶红外光谱-振动圆二色谱仪(FTIR-VCD)和量子化学计算方法对氧化槐果碱的构象结构进行研究。 通过实验与理论计算的结合证明氧化槐果碱分子的优势构象以及相应构象的玻尔兹曼分布比例。
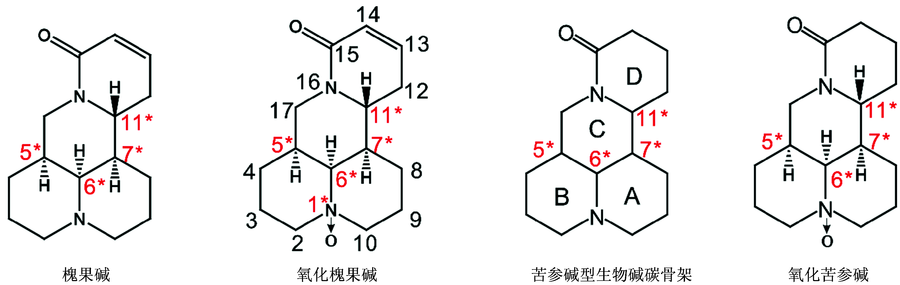 | 图1 苦参型生物碱碳骨架以及氧化槐果碱和槐果碱的化学结构(碳手性中心用* 标明)Fig.1 Matrine framework and chemical structure of sophocarpidin and sophocarpin (* indicates the carbon chiral center) |
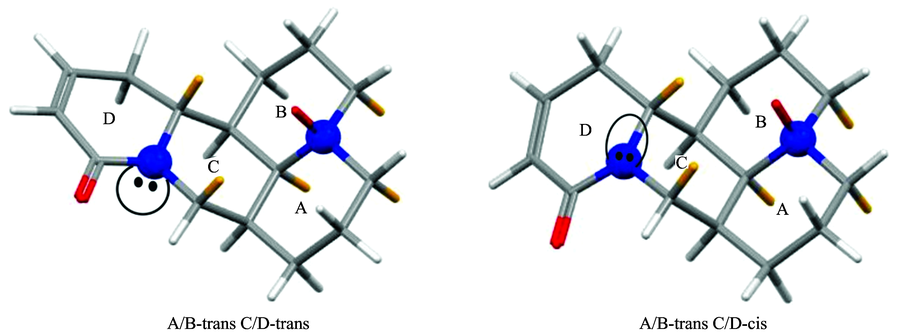 | 图2 氧化槐果碱两种构象Fig.2 Two dominant conformers of sophocarpidin |
氧化槐果碱(HPLC> 98%)购买于成都必应生物技术有限公司(CHENGDU MUST BIOTECHNOLOGY CO, LTD)。 氘代氯仿(CDCl3)购买于Sigma Aldrich 。 Bruker Vertex 70型分光仪和PMA 50 组件(德国BRUKER公司)用于红外光谱和振动圆二色谱的测量。 在整个测量过程中光弹调制器均被设定于1 400 cm-1, 信号分辨率为4 cm-1, VCD光谱在单次扫描1 h, 重复扫描三次(3 × 1 h)的条件下由液氮冷却的MCT检测器获得。 光谱测量范围为1 800~1 100 cm-1。
样品制样时, 5 mg氧化槐果碱样品溶于1 mL氘代氯仿中, 将氧化槐果碱溶液滴在CaF2的表面, 在室温干燥环境下, 氧化槐果碱溶液自然干燥。 溶液的浓度以及垫片的厚度均被反复试验优化, 以保证红外光谱的吸光率在0.2~0.9之间。
在手性分子的构型与构象确认中, 密度泛函算法(DFT)被广泛用于计算VCD光谱[18], 在诸多计算水平中, B3LYP/6-311+G(d, p)和B3LYP/cc-pVTZ计算水平能提供吻合度更高的理论光谱[19]。 本研究采用密度泛函(DFT)方法分别在B3LYP/6-311+G(d, p)和B3LYP/cc-pVTZ水平上对分子的几何结构进行优化并对优化得到的分子构型再进行振动频率分析, 结果无虚频, 表明优化结果确实为稳定构型。 以优化得到的稳定构型为基础, 在相同方法和基组水平下进行VCD光谱计算, 在半高半峰宽为4 cm-1的劳伦兹线型下模拟分子红外吸收光谱和振动圆二色谱, 其频率校正因子为0.98。 所有计算均采用 Gaussian 09程序包[20]完成。
槐果碱具有四个六元环(如图1所示), 其分子中含有C-5, C-6, C-7和C-11四个碳原子手性中心以及N-1和N-16两个氮原子手性中心。 蔡晓丽等提出槐果碱的构型为SSSR(分别对应C-5, C-6, C-7和C-11)[4]。 氧化槐果碱为槐果碱的氧化产物, 它们的C-5, C-6, C-7和C-11构型应该是相同的(如图1所示)。 张越非等指出虽然苦参碱的C-5, C-6, C-7和C-11构型(SSSR)和氧化苦参碱的构型(图1)是相同的, 然而由于N-1上氧原子使得氧化苦参碱的手性构型命名变为SSRR[9] 。 按照同样的手性构型命名规则, 氧化槐果碱分子中A/B环上N-1由于氧原子的作用使得A/B-trans(A/B-cis)中N-1手性为R(S)。 本工作, 氧化槐果碱的理论计算构型均采用SSRR(分别对应: C-5, C-6, C-7和C-11)、 N-1手性为A/B-trans(A/B-cis): R(S), N-16手性为C/D-trans(C/D-cis): R(S)。
氧化槐果碱可能存在的四种构象: A/B-trans C/D-trans(tt), A/B-trans C/D-cis(tc), A/B-cis C/D-trans(ct)和A/B-cis C/D-cis(cc)。 经模拟优化后发现: A/B-trans具有椅型-椅型构象; A/B-cis具有扭船型-椅型构象; C/D-trans和C/D-cis均为椅型-沙发型构象。 对氧化槐果碱四种构象在室温下基于相对能Δ E以及自由能Δ G进行玻尔兹曼分布计算, 发现A/B-cis即顺式连接的构象占比可以忽略不计, 如表1所示, A/B-trans构象的玻尔兹曼分布百分比要远远大于A/B-cis构象。 这个观察结果可通过分析优化后的氧化槐果碱四种构象的结构参数进行佐证。 氧化槐果碱中A/B环的C— H键可粗略分为两种: 一种是平行于桥头氮原子N— O键方向上并和A/B环面大致垂直的直键(axial); 另一种是垂直于桥头氮原子N— O键方向并伸向A/B环外的平伏键(equatorial)。 构象A/B-trans C/D-trans和A/B-trans C/D-cis的二面角θ (Hn, e-Cn-Cn+1-Hn+1, a或Hn, a-Cn-Cn+1-Hn+1, e, 其中Hn, e 为Cn上的直键氢, Hn+1, a为Cn+1上的平伏键氢)都接近60° , 任意相邻两碳原子之间均为邻位交叉, 所以 A/B-trans构象以邻位交叉构象的形式存在, 同时在A/B-trans两种构象中相邻碳上的直键氢与平键氢的距离均大于两氢原子的范德华半径之和, 不存在排斥力, 而A/B-cis C/D-trans和A/B-cis C/D-cis构象中θ 1, θ 2的角度均与60° 相差较大(表2所示A/B环中部分相邻碳之间的二面角以及直键氢与平伏键氢之间距离的数据), 且A/B-cis两种构象中相邻碳上一部分的直键氢与平键氢的距离r(Hn, e…Hn+1, a)或r(Hn, a…Hn+1, e)在230~238 pm之间, 如r1, r2, r3和r4, 它们均小于两个氢原子的范德华半径之和240 pm, 故在A/B-cis构象中, 直键氢与平键氢之间存在排斥力。 由于A/B-trans两种构象中A/B环以邻位交叉构象形式存在, 且相邻碳上平键、 直键氢之间不存在排斥力, 所以在氧化槐果碱四种构象中A/B-trans C/D-trans和A/B-trans C/D-cis两种构象要比A/B-cis C/D-trans和A/B-cis C/D-cis两种构象更稳定。 所以氧化槐果碱的四种构象的结构数据间接证明了表1的数据结果: A/B-trans构象的玻尔兹曼分布百分比要远远大于A/B-cis构象, 即氧化槐果碱可能以A/B-trans C/D-trans和A/B-trans C/D-cis两种构象同时存在(为了简单起见下文均用C/D-trans和C/D-cis表示两种构象), 为实验证明两种构象是否同时存在, 需要将C/D-trans和C/D-cis两种构象的IR和VCD理论计算光谱分别与实验光谱进行对比。
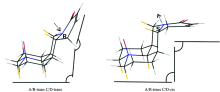
 | 图3 氧化槐果碱的四种构象异构体在B3LYP/6-311+G(d, p)水平上模拟优化后的立体结构Fig.3 The stereochemical structure of four conformers of sophocarpidin optimized at B3LYP/6-311+G(d, p) level |
| 表1 氧化槐果碱不同构象在气相模型中基于相对能Δ E和吉布斯自由能Δ G的玻尔兹曼分布 Table 1 The simulated Boltzmann Population percentage of Sophocarpidin in the gas phase for relative energy (Δ E) and free energy (Δ G) |
| 表2 氧化槐果碱四种构象异构体A/B环上相邻碳上直键氢与平键氢之间的距离以及其二面角(氢原子序号以及二面角在图3中标明) Table 2 The distance between equatorial and axial hydrogen of neighboring CH bonds and the dihedral angle |
图4为氧化槐果碱的主要两种构象异构体C/D-trans和C/D-cis在B3LYP/6-311+G(d, p)水平上的模拟计算光谱图。 其理论的光谱的频率均使用校正因子0.98。 首先查看IR谱图。 如图4所示, C/D-trans(cis) 构象主要吸收峰用a'(a″), b'(b″)等来标记。 IR谱图中, C/D-trans和C/D-cis两种构象的IR光谱及其相似, 而两种构象的VCD谱图大不相同, VCD谱图清晰地显示各个官能团伸缩振动的倾向。 利用Gaussian软件确认氧化槐果碱两种构象VCD图谱中各个吸收峰的来源: 吸收峰a', b', a″和b″分别显示-/+/+/-, 均为C/D环中C13=C14-C15=O振动所引起; 吸收峰c'和c″在VCD谱图中有较大的差异, 吸收峰c'是较宽的正峰, 而吸收峰c″为+/-, 它们均主要由于C-8, C-10, C-12上CH2剪式振动以及C/D环上C— N键的伸缩振动引起, 其中C/D环上C— N键的伸缩振动是引起吸收峰差异的关键, C/D环上C— N键的伸缩振动在吸收峰c'(即C/D-cis构象)的VCD上显示负峰, 而C/D环上C— N键的伸缩振动在吸收峰c'(即C/D-trans)却显示正峰; 吸收峰d'和d″在VCD图谱中均显示正峰, 其中C/D-trans构象中的d'主要由C/D环中C-12的CH2键剪式振动所引起的, C/D-cis构象中的d″则主要由C-12, C-2, C-8和C-10上CH2键的剪式振动所引起的; 吸收峰e'和e″均为负峰, C/D-trans构象中的e'峰主要由C/D环α -C上C— H键的摇摆振动以及C— N键的伸缩振动所引起, C/D-cis构象中e″峰则主要由C/D环上C— H键的剪式振动以及C— N键的伸缩振动所引起; 吸收峰f'和f″均为正峰, 然而C/D-trans构象中f'峰的吸收强度约10倍强于C/D-cis构象的f″峰, 均由C/D环上C— H键扭曲振动所引起; 吸收峰g'和g″均为负峰, C/D-trans构象中g'峰是由A/B/C/D四环上C— H键的扭曲振动所引起, 而C/D-cis构象中g″峰则主要是由C/D环上α -C上C— H键的扭曲振动所引起; 吸收峰h'和h″均显示正峰, 均主要由C/D环上的C— H键的扭曲振动所引起。
 | 图4 氧化槐果碱的两种主要构象异构体的IR和VCD理论谱图与实验谱图的对比Fig.4 The comparison of simulated IR and VCD spectra with the measured spectra for two dominant conformers of sophocarpidin |
氧化槐果碱的两种构象的IR理论光谱差别不大, 而在VCD光谱中, 两种构象的差异极大。 这是由于IR光谱的吸收峰强度与样品分子的电偶极矩的平方(|
| 表4 氧化槐果碱的两种构象C/D-trans和C/D-cis的C/D环上二面角对比 Table 4 The comparison of dihedral angle between two dominant conformers of sophocarpidin |
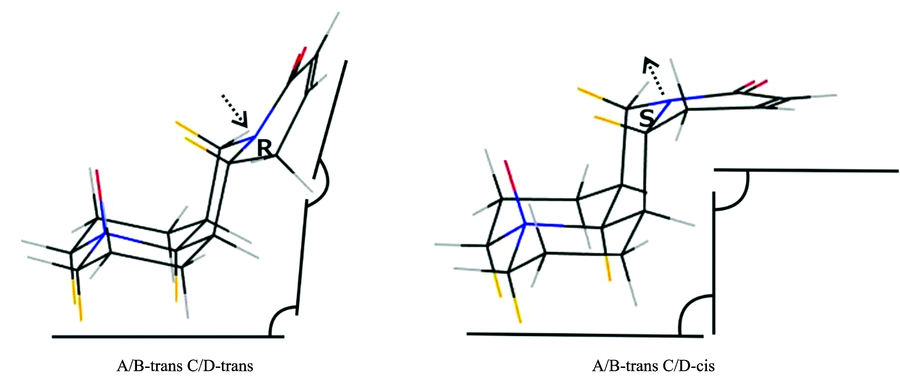 | 图5 氧化槐果碱两种构象环面结构对比图图中箭头表明N-16孤对电子对的方向, 直线与角度分别表示环平面以及环平面之间的角度Fig.5 The comparison of the angle of planar ring between two dominant conformersThe arrows indicates the direction of lone pair of the atom N-16; the line indicates planar ring, the arc indicates the angle between planar rings |
将理论光谱与实验光谱进行比较, 如图4所示, 理论计算的IR光谱基本上与实验一致, 虽然理论模拟光谱的吸收峰强度在某些IR频率区相对实验吸收峰强度较弱。 特别是在1 140 cm-1处主要由C/D环中C— N键振动所引起的吸收峰与1 100 cm-1处的N— O键的吸收峰, 测得的吸收峰强度明显高于理论计算值。 从图中分析可知, 一方面在1 700~1 600 cm-1区域间, 实验VCD光谱是不可能用C/D-trans构象或C/D-cis构象独单独来解释的; 另一方面, C/D-trans和C/D-cis两种构象的VCD理论计算光谱的各个吸收峰均与氧化槐果碱的VCD实验光谱的吸收峰相对应。 从谱图中分析可知氧化槐果碱可能存在着C/D-trans和C/D-cis两种构象。 为了进一步分析氧化槐果碱是否同时存在两种不同构象, 需要根据表1的玻尔兹曼数据计算氧化槐果碱的玻尔兹曼权重理论光谱。
图6是氧化槐果碱在气相模型中, B3LYP/cc-pVTZ和B3LYP/6-311+G(d, p)水平上的理论光谱根据表1玻尔兹曼分布数据而得玻尔兹曼权重分布谱图与实验光谱的对比图。 图中的理论光谱经由波尔兹曼权重分布处理, 所有理论光谱的频率均使用校正因子0.98。 IR谱图中, B3LYP/cc-pVTZ和B3LYP/6-311+G(d, p)水平上的IR理论计算光谱大致相同, 与薄膜环境中进行实验得到的IR实验光谱的波峰数基本吻合。
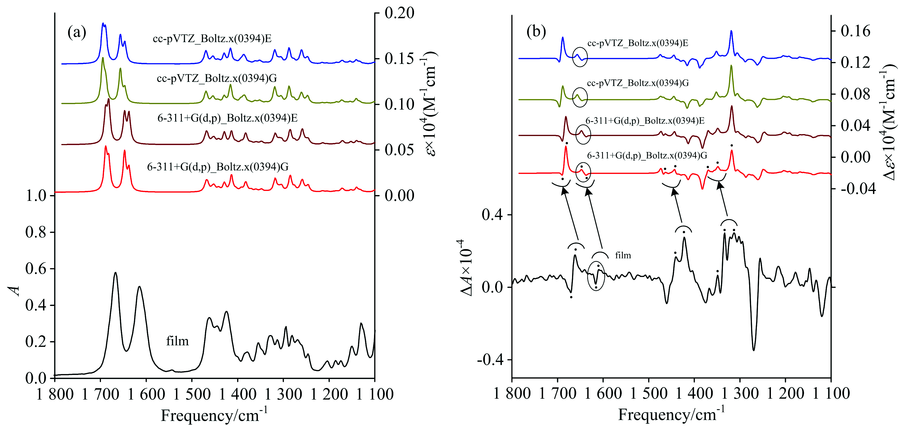 | 图6 氧化槐果碱在B3LYP/cc-pVTZ和B3LYP/6-311+G(d, p)水平上且基于相对能Δ E和吉布斯自由能Δ G的波尔兹曼权重谱图与实验谱图的对比Fig.6 Comparison of the Boltzmann-weighted simulated IR and VCD spectra at B3LYP/cc-pVTZ and B3LYP/6-311+G(d, p) levels for relative energy (Δ E) and Gibbs free energy (Δ G) with the measured spectra |
在VCD谱图中, 在频率范围1 700~1 600 cm-1中C=C与C=O双键伸缩振动区, B3LYP/cc-pVTZ和B3LYP/6-311+G(d, p)水平上的VCD理论计算光谱均为-/+/+/-, 而VCD实验光谱为-/+/-/+。 利用图4分析可知这是由于C/D-trans构象中b'峰的理论计算稍微偏向高频; 在频率范围1 450~1 400 cm-1, C— H弯曲键振动区, VCD实验光谱为+/+, 在B3LYP/6-311+G(d, p)水平计算条件下的理论光谱为+/+, 而 B3LYP/cc-pVTZ水平计算条件下的理论光谱为+, 且在频率范围1 350~1 300 cm-1, VCD实验光谱为+/+/+, 在B3LYP/6-311+G(d, p)水平计算条件下的理论光谱为+/+/+, 而 B3LYP/cc-pVTZ水平计算条件下的理论光谱为+/+。 其余频率范围内, 虽然部分理论光谱出现较弱的峰, 但实验光谱与理论光谱基本相符。 可见B3LYP/6-311+G(d, p)水平上的理论计算能更好地运用于氧化槐果碱的模拟。
在B3LYP/6-311+G(d, p)水平上, 基于吉布斯自由能(Δ G)的玻尔兹曼权重理论光谱与基于相对能(Δ E)的玻尔兹曼权重理论光谱的不同之处在于峰型与峰的强度。 由图6可知, 在1 700~1 600 cm-1区域中, 基于Δ E的IR玻尔兹曼权重分布光谱出现明显的双峰, 而基于Δ G的玻尔兹曼权重分布的双峰要弱很多, 更接近于实验光谱的峰型。 同时, 在1 700~1 600 cm-1区域间C=C与C=O双键伸缩振动区域的VCD光谱中, 基于Δ G的玻尔兹曼权重分布光谱在~1 660 cm-1处有更强的负峰, 更接近于实验光谱。 1 500~1 100 cm-1区域基于Δ G和Δ E的玻尔兹曼权重分布光谱与实验光谱的峰型以及吸收峰数量一致。 相对于两种构象的理论光谱, 玻尔兹曼权重光谱有效提高了理论与实际的相似度, 基于吉布斯自由能(Δ G)的玻尔兹曼权重光谱能更好的反应氧化槐果碱的构象分布。
通过傅里叶红外光谱和振动圆二色谱的实验光谱与理论计算光谱对比, 以及对玻尔兹曼分布数据和玻尔兹曼权重分布图谱分析, 可得出氧化槐果碱以一定比例共存于两种构象异构体: A/B-trans C/D-trans和A/B-trans C/D-cis。 氧化槐果碱各环的构象分别为: A-椅式、 B-椅式、 C-椅式、 D-沙发式, 且两种构象中A/C环与B/C环的连接方式均为B/C-cis, A/C-cis, 六个手性中心分别为: 四个碳原子手性中心5S 6S 7R 11R和两个氮原子手性中心: A/B环上N-1的手性为R, C/D环构象C/D-trans(C/D-cis)中N-16的手性为R(S)。 通过对氧化槐果碱在B3LYP/cc-pVTZ和B3LYP/6-311+G(d, p)的模拟计算, 在不同构象的理论光谱与实验光谱的对比以及在室温条件下的玻尔兹曼权重分布计算光谱与实验光谱的对比中发现: 基于自由能(Δ G)对B3LYP/6-311+G(d, p)基组水平上的模拟计算得到的理论光谱进行玻尔兹曼权重分布更接近氧化槐果碱的实验光谱, 能更好的反应氧化槐果碱的构象分布。 由玻尔兹曼分布数据可知在室温下氧化槐果碱的两种构象A/B-trans C/D-trans和A/B-trans C/D-cis以60%:40%的比例同时存在。
致谢: 加拿大阿尔伯塔大学和加拿大自然科学与工程委员会的经费资助, 同时感谢加拿大西部研究网络(Western Canada Research Grid)和加拿大共享学术研究计算网络(Shared Hierarchical Academic Research Computing Network)对本实验的理论研究提供计算平台; Yunjie Xu教授为加拿大手性和手性识别首席科学家。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|