{kind=link}
基于非谐力场的HOI分子振动光谱研究
[陈恒杰1  , 王全武
, 王全武1 , 张家伟1 , 望军2 ]
, 王全武|
|


作者简介: 陈恒杰, 1980年生, 重庆科技学院数理学院副教授 e-mail: nwwolfchj@gmail.com
应用包含非迭代三激发(CCSD(T))、 迭代三激发(CC3)及其变体(CCSDT-3)的耦合簇理论(CC)和密度泛函理论(DFT)之B3LYP方法, 使用直到五阶的自洽相关一致基组aug-cc-pV
The structure of the HOI molecule was optimized at first, by employing the coupled cluster theory (CC) with single, double, and perturbative triple excitations (CCSD(T)), the iterative triple excitations (CC3) and its variants (CCSDT-3), and the B3LYP method from the density functional theory (DFT) in conjunct with the Dunning’s correlation consistent basis sets aug-cc-pV
活性卤素分子对臭氧消耗具有重要作用, 目前, 人们对卤素氢氧化物HOCl, HOBr与臭氧的反应机理已有深刻认识[1], 最近人们意识到由HOI导致的臭氧消耗要远大于HOCl, HOBr[2]。 Ogilvie等最初在10 K左右的固态氩基质中, 注入碘甲烷和氧气, 发现有HOI存在[3], 随后, Walker等利用氮/氩基质保护使臭氧与HI反应, 据此获得了HOI中间体并解析出其红外光谱[4], Barnes及合作者在298 K和1 013 mbar下光解含碘混合物, 首次报道了HOI在600~4 000 cm-1区间内的气相红外光谱[5], 1996年, Klaassen等对HOI的旋转结构进行了分析[6], 直到2004年, Ozeki等应用微波谱对HOI/DOI在320~670 GHz的纯旋转谱进行了测量, 部分光谱常数和振动平均结构被演绎[7]。 由上可见对HOI的实验研究还远不够透彻, 主要归咎于HOI的瞬态特性和生成的复杂性, 前者导致获取HOI光谱十分不易, 后者则增加了其光谱辨认和解析的难度, 使得HOI的实验光谱数据极其匮乏, 特别在高分辨振转光谱方面。 理论方面, 传统的量子理论在计算振动光谱时均采用谐性近似, 因此获得的频率与实验基频往往有较大差异, 有时甚至给光谱归属造成误导。 此外, 谐性近似未能有效考虑振动激发的非谐效应, 因此只能获得0-1跃迁, 对振动光谱的和频、 倍频预测则无能为力。 近年来, 多个小组在振动谱的非谐效应上取得重大突破, 先后有振动自洽场方法(VSCF)[8]、 振动组态相互作用方法(VCI)[9]和振动二阶振动微扰理论(VPT2)[10]被发展。 针对HOI的非谐振动情况, 就我们所知, 目前仅有Antonio等对其全局势能面进行研究时得到十个振动频率[11]。 综上可见, 无论实验还是理论, 有关HOI的多个振转光谱数据依然是空白的。 因此, 一个精确的理论计算显的十分必要。
应用包含迭代三激发的耦合簇理论CC3及其变体CCSDT-3、 包含非迭代三激发的耦合簇理论CCSD(T)以及密度泛函理论之B3LYP方法优化出HOI分子的基态结构, 接着在平衡结构附近展开微扰获得二阶力场和谐振频率。 继续考虑非谐效应, 沿分子简正坐标数值差分二阶力场, 获得三阶和准对角化四阶力场, 在此基础上分别应用VSCF, VPT2和VCI获得非谐性常数和非谐振动频率。 CCSD(T)计算时H和O采用aug-cc-pVxZ(x=T, Q, 5)基组, I使用考虑相对论效应的赝势基组aug-cc-pVxZ-PP; 综合考虑理论水平和计算成本, CC3与CCSDT-3计算时仅采用aug-cc-pVTZ及对应的aug-cc-pVTZ-PP基组, DFT计算则采用aug-cc-pV5Z及aug-cc-pV5Z-PP基组, 为表示方便, 相应基组缩写为AxZ(x=T, Q, 5)。 CCSD(T)结果由Molpro[12, 13]和CFOUR[14]程序包分别独立获得, 以示区别, 下文标星号者表示Molpro计算结果, CC3和CCSDT-3计算则由CFOUR单独执行, DFT部分由Gaussian09[15]完成。
精确的平衡结构是获取分子光谱常数和振动频率的基础, HOI结构见图1, 本文计算值、 文献以及可获得的实验结果见表1。 Klaassen等通过分析HOCl和HOBr精确转动光谱获得的平衡结构数据, 敏锐的发现两分子的rOH键都收敛于0.964 3 Å [16, 17], 因此, 他推测, 系列化合物HOI的rOH键也应收敛于此。 Ozeki等通过拟合182个HOI和91个DOI的转动谱线得到HOI/DOI的转动常数, 有效分子结构r0被一并计算, 利用获得的离心畸变常数和振动频率, 他们推导出HOI的谐性力场, 进一步考虑了非谐和基质效应后估计了振动平均结构rz和平衡结构参数r0, 本文计算值与上述实验结果非常吻合, 多种方法获得的rOH键长一致收敛于0.964 Å 。 最大误差仅为0.001 7 Å (Molpro的CCSD(T)/ATZ值)。 针对ROI, 实验和多数计算均显示其键长约为1.99 Å , 文献[11]利用CASPT2给出的值最小为1.968 8 Å , 意外的是, 利用CFOUR计算的CCSD(T)/AQZ值仅有1.975 Å , 这与同级别的Molpro结果1.996 Å 相差多达0.021 Å , 类似的情况也发生在小基组ATZ上, 然而当基组进一步增大至A5Z时, Molpro和CFOUR结果完全一致, 这可能由基组不完备和内部参数设置差异引起。 可以看出, B3LYP/A5Z和基于Molpro的CCSD(T)/ATZ均高估rOH键长约0.010~0.015 Å ; 另外, 我们也注意到, 结合ATZ基组, CC3和CCSDT-3取得了与CCSD(T)/A5Z一致的结构参数, 这给我们启示, 鉴于资源限制采用较小基组时, CC3和CCSDT-3在获取分子精确的结构参数上更有优势。
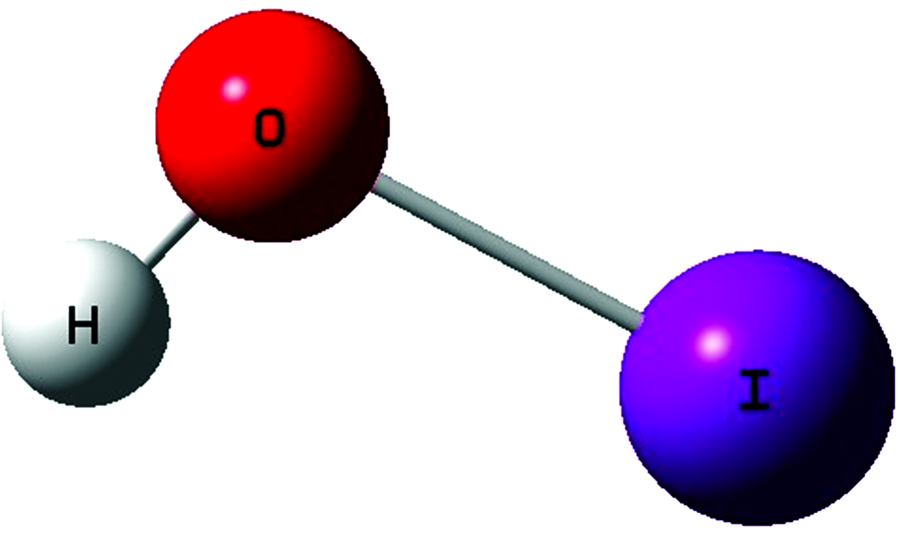 | 图1 HOI分子结构Fig.1 The equilibrium geometries of HOI |
| 表1 HOI分子平衡结构 Table 1 Equilibrium structure of HOI |
基于上述平衡结构, 在其周围展开微扰计算, 先利用解析导数获得谐性近似下的振动频率, 接着数值差分解析二阶力场获得非谐力场, 我们得到了简正坐标下分子的半经典四阶力场, 鉴于篇幅, 详细参数这里不再列出, 基于该力场, 通过VPT2获得了HOI/DOI的振转光谱数据, 具体见表2— 表5。
| 表2 转动常数(MHz) Table 2 Rotational constants (MHz) |
| 表3 旋振相互作用常数(× 10-3 cm-1) Table 3 The vibrational-rotational interaction constants (× 10-3 cm-1) |
| 表4 非谐性常数(cm-1) Table 4 Anharmonic constants (cm-1) |
| 表5 S约化下的离心畸变常数(cm-1) Table 5 S-reduced centrifugal distortion constants (cm-1) |
表2列出了HOI/DOI的平衡转动常数(Ae, Be, Ce)和基态转动常数(A0, B0, C0), Klaassen等的红外观测实验显示, HOI的A0, B0, C0分别为20.934 75, 0.278 939 8和0.275 070 5 cm-1, 将其统一折算到MHz单位后为627 608, 8 362.4以及8 246.4, 相应Ozeki的纯转动实验结果为627 757.4, 8 366.2和8 246.4 MHz, 由表2可以看出, 应用耦合簇方法计算的基态转动常数与实验结果非常吻合, 特别是CCSDT-3/ATZ, 其计算的A0与两组实验间的绝对误差只有98.9和50.5 cm-1, 相对误差仅为0.016%和0.008%, B0的相对误差分别为0.51%和0.47%, C0的两实验结果完全相同, 与CCSDT-3/ATZ值误差为0.49%; 另外, 我们也观察到, 采用CCSD(T)结合AxZ(x=T, Q, 5)计算时, 各转动常数并未随基组增大而趋于某一极限, 很明显CCSD(T)/AQZ值要大于CCSD(T)/ATZ和CCSD(T)/A5Z值, 且有远离实验值的趋势, CCSD(T)/A5Z虽然减少了与实验值间的差异, 但误差还是大于CCSD(T)/ATZ结果, 这反应出转动常数计算并非基组越大越好, 有时候盲目扩大基组不仅不能使计算结果更加接近实验值, 而且会增加计算资源的消耗。 表中数据也显示, B3LYP/A5Z计算的转动常数与实验值差异最大, 这很好理解, 密度泛函在结构优化上显示了巨大的潜力和良好的特性, 但在描述相互作用上有很多不足, 这导致应用B3LYP描述的势能面与“ 真实” 情况差异较大。 对于DOI, 同样CCSDT-3/ATZ结果与Ozeki等的纯转动光谱实验值最接近, CCSD(T)/ATZ, CC3/ATZ次之, CCSD(T)/AQZ, CCSD(T)/A5Z和B3LYP/A5Z偏差较大。
就我们所知, 目前还没有振转相互作用常数的报道, 我们预测的结果列于表3, 可以看出, 各CC间结果差异不大, 但 B3LYP与CC结果差异较大, 特别在
基于Klaassen观测的跃迁, 非谐常数x11的实验值为-82.825 cm-1, 表4列出的CC计算结果均约85 cm-1, 略大于实验值2.1 cm-1, 而文献给出的x11为-99.321, 可能是由于CASPT2考虑动态关联能不足导致势能面不精确引起, 针对x12, CC结果大于文献值2.1倍, 反之x13, 文献结果却是CC值的3.9倍。 对DOI, x11实验值为-43.445 cm-1, CC结果集中于45 cm-1, 与实验符合很好。 除x13外B3LYP计算的非谐性常数与CC相当, 且B3LYP计算值普遍偏小(x22例外), 依据转动常数以及其他已有实验结果比较, 有理由相信当前CC计算结果是更可靠的。
表5列出了对称约化(S约化)下的离心畸变常数, Klaassen等的实验结果显示HOI的DJ为0.235 76× 10-6 cm-1, CC计算的DJ与此非常接近, 特别是CCSDT-3/ATZ, 其结果0.235 6× 10-6 cm-1与Klaassen的实验结果几乎一致, Ozeki等利用纯转动光谱获得的DJ略大为0.257 53× 10-6 cm-1, 大于当前所有计算值, 与CCSD(T)/A5Z结果接近。 针对DK值, 两实验结果基本一致, CC结果总体偏小8%左右, 但B3LYP结果约为实验值的9倍, 进一步说明B3LYP在获取准确光谱常数上具有偶然性, 同理, 对DJK值, 当前CC计算值与实验结果符合很好, B3LYP与实验结果差异多达19倍, 在d1上, CC结果与文献[7]符合较好, 但与Klaassen等人给出的结果为-18.54相差较大, 这或许与Klaassen在拟合时直接将d2固定为零有关, 这样间接抬高了d1的作用, 从d2结果也可看出CC与Ozeki实验结果符合较好, 尽管两者之间存在24.9%的差异, 但这主要是因为d2很小的原因, 从表看出, Ozeki等的纯转动光谱与CC值基本一致, 反应出CC结果的合理性、 正确性。 对于DOI, 仅有一个实验报道, 与CC计算结果符合较好。
HOI/DOI为平面结构, 属Cs点群, 两对称元A'和A″, 三个基频v1, v2, v3都具有A'对称性, 分别表示OI伸缩振动、 HOI弯曲振动和OH伸缩振动, 基于半经典四阶非谐力场, 利用VPT2, VCI和VSCF方法, 我们计算了HOI及其同位素取代物DOI的三个基频和直到v1+v2+v3=3的倍频与和频, 几组可获得的实验和文献值被一同列于表6和表7。 可以看出, 三个基频振动强度较大, 其中OH伸缩振动v3具有最大振子强度, 如利用VPT2/CC3/ATZ得到的振子强度为86.9 km· mol-1, 这表明该振动峰在实验上最容易被发现, 气相和旋振实验显示v3为3 620和3 626 cm-1, 而Ar基质实验结果仅为3 597 cm-1, 主要原因是Ar基质与OH间的相互作用导致OH振动变慢, 本文计算的v3值分布于3 599~3 640 cm-1, 其中VSCF最小为3 599 cm-1, 使用CFOUR程序包的VPT2/CCSD(T)/AQZ给出的结果最大, 其值为3 645 cm-1, 其他方法与旋振实验结果的差异均小于20 cm-1, 特别是VPT2/CCSD(T)/ATZ结果, 与气相结果完全一致, 与旋振光谱数据仅有6 cm-1差异。 同时我们也注意到, 基于相同的方法CCSD(T)和基组AxZ, Molpro与CFOUR计算结果有轻微差异, 具体到v3, Molpro计算值总小于CFOUR值而更接近于实验值, 如CFOUR在VPT2/CCSD(T)/ATZ下给出的v3值为3640 cm-1, 而Molpro给出的结果为3 620 cm-1, 两者相差多达20 cm-1, 造成该差异的原因可能来自于以下三方面: (1) Molpro计算时采用冻结核近似, 而CFOUR默认全电子计算, 理论上CFOUR计算的势更准确, 但经验显示全电子基组有时会导致过渡校正现象, 即基组越大振动频率与实验值偏差反而增大; (2) 两程序采用的收敛标准、 微扰时格点间距、 势能面取值范围等参数有所不同也会造成轻微的差异; (3) 势能面拟合时Molpro采用完全四阶多项式, CFOUR使用半经典四阶多项式, 其基函数要少的多, 这点可从二者给出的简正坐标力场数目清晰的看到。 在VPT2/CCSD(T)/AxZ(x=T, Q, 5)理论水平下, Molpro给出的v1位置依次为572, 580和582 cm-1, 相应v2为1 081, 1 085和1 086 cm-1。 三个v1实验结果集中于575 cm-1附近, 除Ar基质外, 其余实验显示v2带中心在1 070 cm-1, 我们观察到, 随基组增加, Molpro结果具有良好的自洽收敛特性, 因此对其进行完全基组外推极限(CBS)是没有问题的, 反过来也并非基组越大越好, 至少在v1, v2, v3三个基频振动上基组越大, 相应计算值与实验结果差异反而变大, 恰恰最好的结果来自于各类方法与ATZ基组结合获得的计算值, 进一步说明, 非谐计算时, 不能盲目地通过增大基组来预期改善振动频率, 否则不仅浪费了计算资源, 还存在结果变得更“ 糟” 的风险。 同样情况下CFOUR给出的v1值为 584, 592和588 cm-1, v2为1 071, 1 068和1 072 cm-1, 可以看出, 随基组增大CFOUR计算的振动频率大小出现了涨落现象, 使得利用幂函数衰减方式将振动频率推导到CBS极限变得不再可靠, 一个可能的原因是本文采用的EMSL基组大小一致问题。 为了比较, 本文也应用VPT2/B3LYP/A5Z进行计算, 意外的是, 尽管VPT2/B3LYP/A5Z计算的某些光谱常数不尽人意, 但由其获得的振动频率与实验结果符合的非常好, v1, v2, v3值分别为575, 1 066和3 629 cm-1, 该结果甚至比本文使用的多类耦合簇结果更接近实验值, 这反应出计算非谐振动频率结果不一定耦合簇最佳, 有时密度泛函、 微扰理论也可得到优秀的结果; 通过与三个基频振动和v3的倍频振动实验结果比较, 我们注意到VCI结果要好于VSCF和VPT2, 同样ATZ基组下, VSCF得到的结果最差。 由于HOI/DOI的瞬态和偶极特性, 大部分振动频率未能被实验观测, 我们预期的频率可能为将来实验探测提供帮助。 整体来看, Molpro软件下VCI/CCSD(T)/ATZ, CFOUR软件下VPT2/CC3/ATZ与Gaussian软件下VPT2/B3LYP/A5Z的计算结果与已知实验结果符合最好, 据此我们推断v1, v2的倍频出现在1 135和2 130 cm-1, 其中v2与v3具有相似的强度, 可能在将来实验中观测到, 但由于v2与2v1相差仅80 cm-1, 且v2强度远大于2v1, 在低分辨谱中, 2v1可能被v2掩盖, 另外, 三个和频v1+v2, v1+v3, v2+v3带具有较弱的强度0.06, 0.22和0.69, 需在高浓度、 高灵敏光谱仪下被观测, 其位置约在1 650, 4 205和4 685 cm-1附近。
| 表6 HOI的基频、 倍频与和频 Table 6 Fundamental, Overtones and Combination of HOI |
| 表7 DOI的基频、 倍频与和频 Table 7 Fundamental, Overtones and Combination of DOI |
应用耦合簇理论方法CCSD(T), CC3, CCSDT-3和密度泛函方法中的B3LYP方法, 结合VSCF, VPT2和VCI研究了基于四阶力场的HOI非谐光谱常数、 振动频率和振子强度, 基组效应、 氘取代效应。 与已有的实验结果比较表明: 耦合簇理论获得的HOI/DOI分子常数是可靠的, 特别是迭代三激发CC3/ATZ, CCSDT-3/ATZ, 其获得的光谱常数与实验值最接近, 尽管B3LYP在有些分子上取得了良好的结果[18, 19, 20], 但在HOI/DOI上, 其计算的光谱常数与耦合簇及实验结果存在较大偏差, 因此应用DFT预测光谱常数时要特别小心, 尽管如此, B3LYP计算的非谐振动频率却与耦合簇和实验结果非常一致; 同时我们也看到, 并不是基组越大, 结果越接近实验值, 针对耦合簇方法, ATZ基组似乎是个不错的选择。 基于上述可靠的判断, 我们预测了HOI若干未知的分子常数, 如HOI下一个最可能观测的振动形式为弯曲振动的倍频2v2, 其位于~2 137 cm-1, 对应DOI为~1 550 cm-1处。 本文计算将为解释和观测HOI/DOI振动光谱提供重要依据。
致谢: 感谢重庆科技学院化学化工学院陈双扣教授提供Gaussian软件的计算帮助。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|