

{kind=link}
{kind=link}
{kind=link}
{kind=link}
碱金属团簇(Li2F) nM (M=Li, Na, K; n=1, 2)结构、稳定性和红外光谱的理论研究
[李小军 , 贺仙梨, 宋瑞娟]
, 贺仙梨, 宋瑞娟]
, 贺仙梨, 宋瑞娟]
|
|


作者简介: 李小军, 1981年生, 西安文理学院化学工程学院副教授 e-mail: xjlicms@yahoo.com
利用密度泛函理论B3LYP方法和6-311+G*全电子基组对(Li2F) nM (M=Li, Na, K; n=1, 2)团簇的几何结构进行了优化, 确定了它们的基态结构, 并对它们的化学稳定性、 电子特性和红外光谱进行了理论研究。 结果表明: (Li2F)M (M=Li, Na, K)团簇具有相似的双三角基态结构, 但是(Li2F)2M的基态结构则完全不同; (Li2F)Li和(Li2F)2Na团簇具有较大的键能和HOMO-LUMO能隙, 致使其具有较高的化学稳定性。 通过轨道分析发现, 这两个稳定团簇的HOMO和LUMO轨道都是由 sp杂化而形成了 σ键。 同时, 也发现(Li2F)2K团簇因具有较低的电离势(4.23 eV), 可以考虑其为新型的超碱金属化合物。 此外, 模拟了(Li2F)nM团簇的红外振动特征峰, 并对主要谱峰的振动模式进行了指认。
Using the density functional theory (DFT), the geometrical structures of the (Li2F) nM (M=Li, Na, K; n=1, 2) clusters were optimized at the B3LYP/6-311+G* level of theory, and all of ground-state structures were determined, while their chemical stabilities, electronic properties and infrared spectra were systematically discussed. The calculated results showed that the (Li2F)M (M=Li, Na, K) clusters had the same double-triangular geometries, whereas the structures of (Li2F)2M were distinctly different. It was also found that the (Li2F)Li and (Li2F)2Na clusters were strongly stable due to the large binding energy and HOMO-LUMO energy gap. The high chemical stability can be explained by the strong sp hybridization to form the σ bonds (e. g., HOMO, LUMO). Meanwhile, it was predicted that the (Li2F)2K cluster could be considered as novel superalkali compound because of its low ionization potential (4.23 eV). In addition, we simulated the infrared spectra of these (Li2F) nM clusters, and assigned the main vibrational peaks for further experimental references.
众所周知, 超碱金属化合物由于具有较低的电离势, 而具有特殊的应用价值, 例如在激光技术、 光通讯、 光存储、 光计算机等方面有着潜在的优势[1, 2, 3, 4]。 在1980年初, 超碱金属吸引了许多科学家的极大关注, 对其进行了大量的实验和理论研究。 超碱金属的概念最初是由Gutsew和Boldyrew[5]提出的。 随后, Wu等[6]采用密度泛函理论方法设计了新型超碱离子化合物, 并且在2014年发现了第一个超碱金属团簇L
近年来, Srivastava等[9]采用密度泛函理论(DFT)方法对LinF (n=2~5)团簇的结构和电子性质做了研究, 发现Li2F团簇具有较低的电离势, 而且研究也表明具有低电离势的化合物同时也会出现较强的二阶非线性光学响应, 可以被应用于非线性光学材料领域。 值得一提的是, 碱金属修饰的团簇结构一直都是非线性材料研究的热点, 例如碱金属修饰的硅锗团簇[4, 10]等, 为新材料的开发和应用提供了重要的研究基础。 碱金属原子对团簇化合物的结构修饰, 将进一步提高团簇化合物在非线性光学上的应用前景。 特别地, 吸附团簇的理论研究有助于我们从分子层面上进一步深入理解碱金属原子与团簇表面相互作用的微观机制。
因此, 采用DFT方法对不同碱金属原子(Li, Na, K)修饰(Li2F)n (n=1, 2)团簇的几何结构和化学稳定性进行了系统研究, 探讨了碱金属原子吸附的不同点位, 以及吸附后各种性质的变化, 例如键能, 前线轨道能隙, 红外振动光谱归属等, 同时研究了碱金属吸附团簇的电荷转移机理和轨道贡献。 尤其是, 通过K原子吸附的(Li2F)2K团簇具有较低的电离势, 并对其红外光谱振动特征峰做了归属, 为此类新型非线性光学材料的制备和性能表征提供了理论参考。
采用gaussian09程序包[11]中的杂化密度泛函B3LYP方法[12], 在全电子基组6-311+G* 级别上对碱金属吸附的(Li2F)nM (M=Li, Na, K; n=1, 2)团簇进行优化计算。 先前研究也表明[13, 14, 15], 杂化DFT-B3LYP方法可以很好的处理吸附团簇表面的相互作用, 同时我们考虑了结构内部的色散力D3校正。 所有获得的几何结构都是在振动频率验证其为势能面上稳定点的基础上, 研究其各种性质, 包括不同的吸附点位、 键能、 电子转移和光谱等。 全部计算均在I7计算机上完成。
碱金属吸附团簇(Li2F)nM (M=Li, Na, K; n=1, 2)的键能(Eb)和能隙(Eg)的计算公式如下
图1给出了在B3LYP/6-311+G* 水平下优化得到的(Li2F)nM (M=Li, Na, K; n=1, 2)团簇的平衡几何构型(对于每种团簇, 只给出了能量最低的基态构型)。
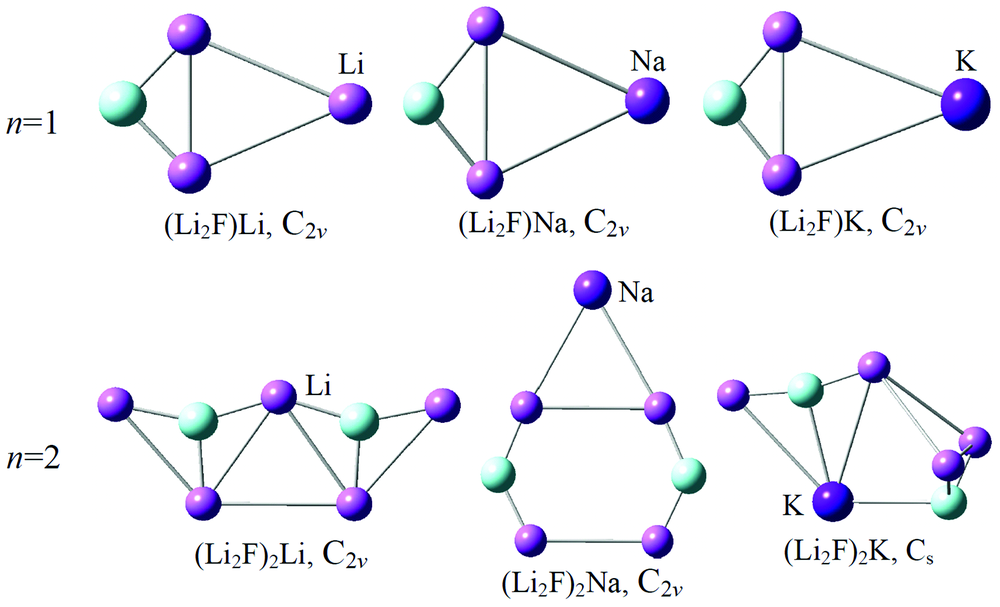 | 图1 优化(Li2F)nM (M=Li, Na, K; n=1, 2) 团簇的基态结构Fig.1 Optimized ground-state structures of the (Li2F)nM clusters |
(Li2F)Li: 该团簇的几何结构对称性为C2v点群。 文献[9]报道发现Li2F为一个等腰三角形。 在该团簇中Li原子吸附在等腰三角形的侧棱Li— Li键上, 形成一个Li— Li— Li桥结构。 三个Li— Li键的键长分别为3.03, 2.50和3.03 Å , 形成一个类似等腰三角形的构型, 其为自旋单重态。 结构参数分析发现, 预测的Li— F键键长为1.70 Å , Li— Li— Li键角48.65° , 最低红外振动频率为165.22 cm-1, 电子态为1A1。 值得一提的是, 对于(Li2F)Li团簇还存在一个稳定的四面异构体, 其F原子吸附在三个Li原子的表面, 形成一个类似三角锥的结构, 但是其能量比基态结构的要高约0.28 eV。
(Li2F)Na: 该团簇的几何构型与(Li2F)Li团簇的类似, 掺杂的Na原子吸附在Li2F等腰三角形的Li— Li侧棱上, 其对称性为C2v点群, 电子态也为1A1。 (Li2F)Na团簇的平均键长比(Li2F)Li的要大, 两个Na— Li键的键长均为3.20 Å , 比(Li2F)Li团簇中的Li— Li键要长0.17 Å 。 Li— Na— Li键角为46.9° , 有所减小, 是因为其增长的Na— Li键导致的, 预测的最低红外振动频率为144.79 cm-1。
(Li2F)K: 根据体系总能量越低几何结构越稳定的原则, 经过优化选取了如图1中的稳定结构, 对称性为C2v。 该团簇同(Li2F)Li和(Li2F)Na类似, 都是掺杂原子吸附在Li— Li键上, 自旋多重态为单重态。 该结构中的两个K— Li键键长分别为3.72和3.73 Å , Li— F键键长为1.69 Å 。 Li— K— Li键角为40.49° , 最低振动频率为107.99 cm-1。 对于(Li2F)K团簇, 同(Li2F)Li团簇一样, 也存在一个稳定异构体, 但是其比基态结构能量高0.31 eV。
(Li2F)2Li: 如图1所示, 此团簇的稳定几何结构具有C2v点群, 自旋多重态为二重态。 对于该结构, 五个Li原子与两个F原子互相连接形成一个状如“ 船型” 的几何结构。 中间Li原子与两个F原子连接所形成的键角F— Li— F为144.29° 。 该团簇的电子态为2A1, 最低振动频率为29.20 cm-1。
(Li2F)2Na: 该团簇的基态结构也具有C2v点群。 几何构型为: 掺杂的Na原子与两个Li原子相连接形成了一个等腰三角形, 而其吸附在一个由Li和F原子组成的六元环上, 类似于“ 金字塔” 型的结构, 此结构特征明显不同于(Li2F)2Li的基态结构。 两个Na— Li键键长都是3.12 Å , 键角Li— Na— Li为60.84° 。 电子态为2A1, 最低振动频率为42.03 cm-1。
(Li2F)2K: 如图1所示, 当K原子吸附在(Li2F)2团簇上时, 其稳定结构明显不同于Li和Na原子吸附的结构。 (Li2F)2K基态结构由一个K原子吸附在Li2F底部, 同时也连接了另一个Li2F中的F原子, 形成了一个Cs点群的平面稳定结构, 其电子态为2A'。 结构上, K原子与两个F原子相连接的K— F键长分别为2.74和2.73 Å , 与两个Li原子相连的K— Li键长则为3.38和3.45 Å , 其F— K— F键角为100.68° , Li— Li— Li键角为59 .97° 。 预测的最低红外振动频率为36.62 cm-1。
2.2.1 键能
根据式(1), 我们对(Li2F)nM (M=Li, Na, K; n=1, 2)团簇的键能进行了计算, 其键能变化曲线如图2所示。 n=1, 其键能分别为2.11, 2.08和2.02 eV (见表1)。 当n=2时, 其键能分别为2.38, 2.40和2.31 eV。 对于M取相同原子, n分别为1和2, 都是n越大, 键能Eb越大。 众所周知, 键能的增加意味着结合一个原子所释放的能量越多, 体系也就越稳定。 如表1所示, 对于n相同, 而M分别为Li, Na和K时, 其键能的变化关系有所不同。 当n=1时, 掺杂不同原子的键能关系为: (Li2F)Li> (Li2F)Na> (Li2F)K; 当n=2时, 则为(Li2F)2Na的键能最大, (Li2F)2Li次之, (Li2F)2K最小。 对于整个团簇体系来说: 键能越大, 团簇结构就越稳定。 所以, 当n=1时, 较稳定的团簇结构为等腰三角形的(Li2F)Li; 然而, 当n=2时, 预测的稳定团簇为(Li2F)2Na平面结构, 正如图1所示。
 | 图2 团簇(Li2F)nM的键能(Eb)、 能隙值(Eg)和电离势(VIP)的变化曲线Fig.2 The curves of the binding energies, energy gaps, and vertical ionization potentials for the (Li2F)nM clusters |
| 表1 团簇(Li2F)nM (M=Li, Na, K; n=1, 2)的键能(Eb)、 能隙值(Eg)、 电离势(VIP)和自然电荷布局 Table 1 The binding energies, energy gaps, vertical ionization potentials and natural populations for the (Li2F)nM clusters |
2.2.2 能隙值
能隙值(Eg)描述了电子从HOMO轨道(最高占据轨道)跃迁到LUMO轨道(最低空轨道)的能力, 也是团簇动力学稳定性的重要指标。 根据式(2), 我们知道能隙值可通过HOMO和LUMO轨道的能量差值来衡量, 即为Eg越大, 则电子从HOMO跃迁到LUMO所需的能量越大, 其化学活性越差, 从而团簇动力学稳定性越高。 从表1可以看出, 对于(Li2F)nM (M=Li, Na, K; n=1, 2)团簇而言, 其HOMO-LUMO能隙值在1.90~2.48 eV之间。 在图2中列出了这些团簇能隙值的变化曲线, 从中我们可以看出能隙峰值出现在2.48 eV, 来自于平面的(Li2F)2Na团簇, 所以该团簇具有较强的动力学稳定性和较低的化学活性。 (Li2F)Li团簇也有较大的能隙(2.34 eV), 所以其化学稳定性也较强。 (Li2F)nK (n=1, 2)两个团簇存在较小的能隙, 分别为1.94和1.90 eV, 表明了这两个团簇的动力学稳定性较差。
2.2.3 轨道分析
为了进一步研究团簇的电子特性, 我们对稳定的(Li2F)Li和(Li2F)2Na团簇的前线分子轨道进行了分析, 同时我们也采用自然键轨道方法对这些前线轨道的原子贡献做了研究。 从图3可知, 团簇(Li2F)Li的HOMO和LUMO分子轨道上的电子云主要分布在Li3团簇上, 与F原子无关。 通过轨道分析可知, HOMO和LUMO分子轨道都是由sp杂化而形成σ 键, 其中HOMO轨道是离域的Li— Li— Li σ 键, 该分子轨道成份为: 84.5% Li-2s和13.5% Li-3p。 然而, LUMO轨道由两个定域的Li— Li σ 键组成, 其轨道成分为: 53.7% Li-2s和43.2% Li-3p。
 | 图3 团簇(Li2F)Li和(Li2F)2Na的HOMO和LUMO轨道Fig.3 The HOMOs and LUMOs of the (Li2F)Li and (Li2F)2Na clusters |
相似地, 团簇(Li2F)2Na的HOMO分子轨道主要来自于s和p轨道的贡献。 其中, HOMO轨道一部分(右侧)来自于Na-3s和Li-2p轨道杂化形成的Na— Li σ 键, 其轨道成份为: 18.9% Na-3s和8.6% Li-2p; 另一部分(左侧)来自于Li原子的2s, 2p和3p轨道杂化而成的Li— Li σ 键, 其成分为46.8% Li-2s, 15.0% Li-2p和7.2% Li-3p。 LUMO分子轨道一部分(右侧)来自于Na原子的3s和3p轨道, 成份为: 33.7% Na-3s和19.7% Na-3p; 另外一部分(左侧)来自于Li原子的19.0% Li-2s和16.3% Li-3p杂化而成的Li— Li σ 键, 其成分为: 19.0% Li-2s和16.3% Li-3p。 这些原子成份贡献将有助于我们进一步理解这些团簇的电子性质, 并为以后这些团簇材料的光电特性提供理论参考。
2.2.4 电离势
超碱金属团簇拥有比碱金属更低的电离势。 然而, 低的电离势可作为优良的非线性光学材料[6, 9]。 我们计算了(Li2F)nM (M=Li, Na, K; n=1, 2)团簇的电离势如表1所示。 结果发现: 与Li原子的电离势(IP=5.39 eV)相比, 团簇(Li2F)Li的电离势较大(5.45 eV), 而团簇(Li2F)Na, (Li2F)K, (Li2F)2Li, (Li2F)2Na和(Li2F)2K的电离势均小于Li原子的电离势。 图2反映了团簇(Li2F)nM的电离势变化趋势, 从图中可以看出, 随着n的增加, 电离势则相应减小。 值得一提的是, 在图2中我们发现团簇(Li2F)2Na的电离势为一个峰值(5.16 eV)。 同时, 除了团簇(Li2F)Mi外, 其他团簇因具有较低的电离势, 尤其是团簇(Li2F)2K (4.23 eV), 可作为潜在的非线性光学材料。
通过自然键轨道(NBO)理论对团簇(Li2F)nM中的基态结构进行自然电荷布局分析, 其M原子的自然电荷和电子构型列在表1中。 从表1中可以看出, 团簇(Li2F)Li中的掺杂Li原子得到了0.14个电子。 从电子构型上可以将Li原子价层2s(1.00)轨道与团簇中Li原子电子构型([core]2s(1.10)3p(0.04))进行比较, 结果发现: 2s轨道得到0.10个电子, 3p轨道得到0.04个电子, 所以该团簇的掺杂Li原子有较强的电负性。 团簇(Li2F)Na与(Li2F)Li的自然电荷布局大体保持一致, 只是电子转移数不同, 例如(Li2F)Na团簇上的Na原子得到0.15个电子。 但是, 对于(Li2F)K团簇而言, 其K原子电子构型为([core]4s(0.94)5p( 0.03))。 明显的是, 其K原子的4s轨道失去0.06个电子, 而5p轨道得到0.03个电子, 所以掺杂的K原子总体失去0.03个电子。
分析发现掺杂Li和Na原子得到电子, 而K原子失去电子, 这应归因于K比Li和Na两个原子活泼造成的。 对于团簇(Li2F)nM言, n=1与n=2时的电荷和电子构型明显不同。 例如, 对于团簇(Li2F)2Li和(Li2F)Li中掺杂的Li原子来说, 电子的得失是相反的, 表明结构特征明显地影响了碱金属的电荷转移方向和数量。 从表1中也可以看出, 团簇(Li2F)2M中掺杂Li和K原子时, 分别失去0.78和0.55个电子。
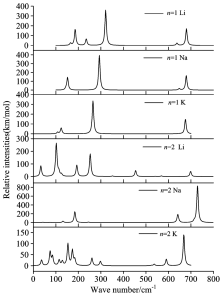
图4给出了在B3LYP/6-311+G* 理论水平上计算得到团簇(Li2F)nM (M=Li, Na, K; n=1, 2)最稳定结构的红外振动光谱。 分析发现, 平面双三角的(Li2F)M (M=Li, Na, K)团簇结构存在几乎相似的红外振动峰。 但是, 对于(Li2F)2M团簇而言, 它们不仅具有不同的红外特征振动峰, 而且其频率和强度发生了明显的变化, 这是因为当碱金属原子(Li, Na, K)吸附在(Li2F)2团簇时, 形成了完全不同的稳定结构(如图1所示)。 从图4中可以看出, 团簇(Li2F)M (M=Li, Na, K)的最稳定结构的红外光谱主要分布在两个区域内: Ⅰ (250~350 cm-1) 和Ⅱ (650~700 cm-1), 它们较强振动峰主要分布在第I区域内。 然而, 团簇(Li2F)2M (M=Li, Na, K)稳定结构的特征振动峰则明显不同。 例如, (Li2F)2Li团簇在低频区(0~300 cm-1)存在四个特征峰, 而(Li2F)2Na和(Li2F)2K在高频区(650~750 cm-1)存在较强的红外振动峰。 在红外振动光谱中, 每条谱峰基本都是由几个振动模式叠加而成, 因此要想指认出各团簇光谱的不同振动模式, 对红外光谱进行归属必须通过理论模拟。
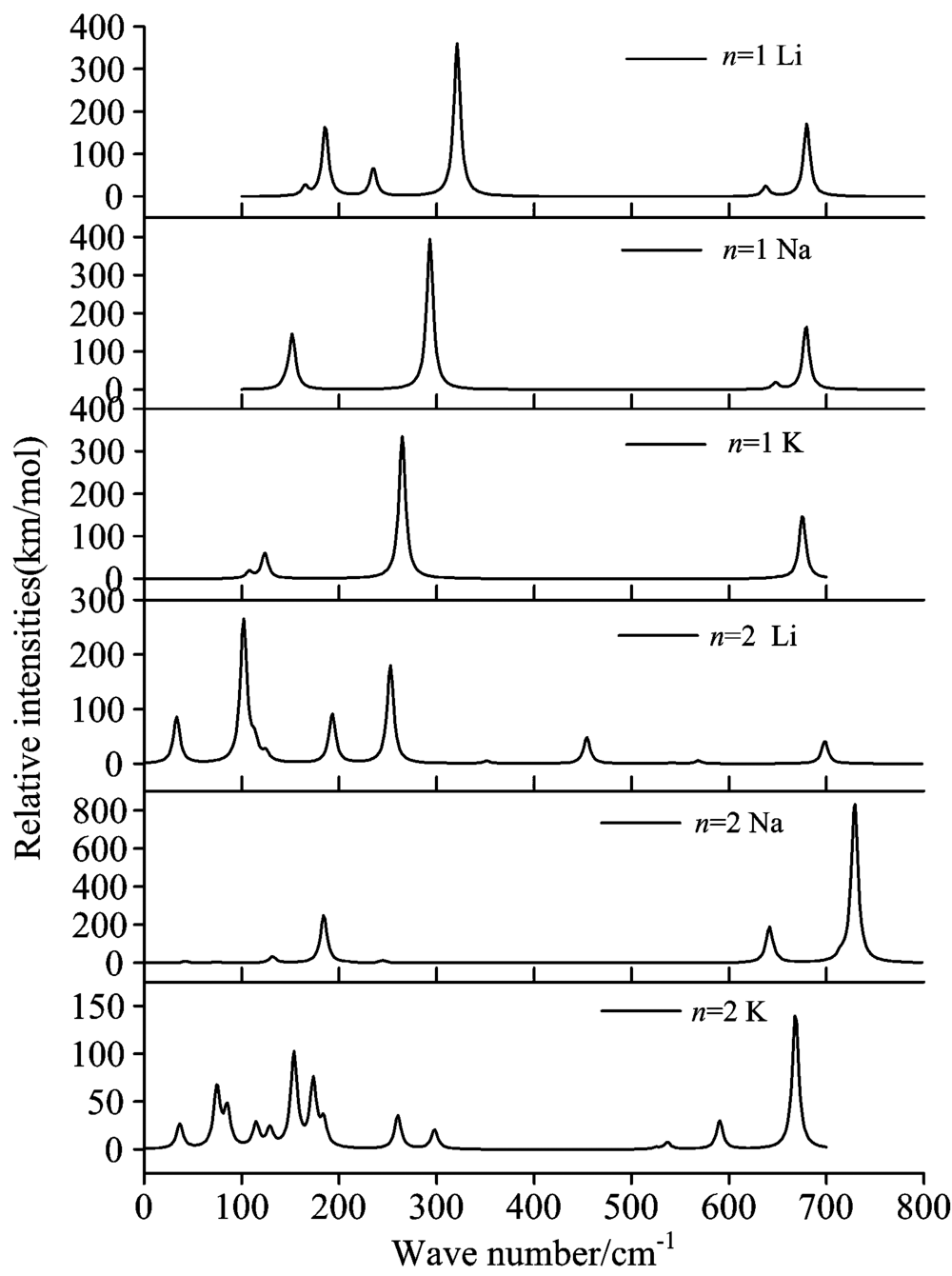 | 图4 团簇(Li2F)nM (M=Li, Na, K; n=1, 2) 模拟的红外光谱Fig.4 Simulated IR spectra of the most stable (Li2F)nM clusters |
(Li2F)Li: 该团簇有三个较明显的振动峰。 321.15 cm-1处是其最强的振动峰, 归属于三个Li原子的对称伸缩振动。 两个中等振动峰: 一个位于波数185.85 cm-1处, 该处振动模式为Li原子的剪式振动; 另一个位于679.81 cm-1处的振动峰, 归属于两个Li原子和F原子的对称伸缩振动。 该团簇的偶极矩为2.68 D。 (Li2F)Na: 对(Li2F)Na团簇来说, 波数为293.19 cm-1处的峰是该团簇最强的振动峰, 其归属于F原子与相邻两个Li原子的剪式振动。 其他两个位于152.08和679.07 cm-1的振动峰分别归属于F原子与两个Li原子引起的摇摆振动和对称伸缩振动。 事实上, 振动峰152.08 cm-1旁边还存在一个弱的振动峰(144.79 cm-1), 其归属于两个Li原子和Na原子的对称伸缩振动。 (Li2F)K: 该团簇有三个振动峰, 分别为123.84, 264.81和675.37 cm-1。 其中, 264.81 cm-1为最强振动峰, 归属于F原子与两个Li原子引起的剪式振动; 次强峰675.37 cm-1归属于F原子与两个Li原子的对称伸缩振动; 位于低频区的123.84 cm-1振动峰, 主要来自于Li— F— Li的摇摆振动。 此团簇的振动峰归属相似于(Li2F)Na团簇。 值得一提的是, 当碱金属(Li, Na, K)吸附在Li2F团簇表面时, 其最强红外吸收峰发生明显红移。
(Li2F)2Li: 对于团簇(Li2F)2Li而言, 它有着比较复杂的振动峰。 在波数102.08 cm-1处为最强的红外峰, 归属于F— Li键的摇摆振动。 位于252.64 cm-1处的次强峰来自于底部Li— Li键的伸缩振动。 位于33.41和193.05 cm-1处的两个中等强度的峰, 分别归属于上部F— Li键的面外摇摆振动和下部F— Li键的平面内摇摆振动。 该团簇的偶极矩较大, 预测为3.15 D。 (Li2F)2Na: 相比较而言, (Li2F)2Na具有简单的红外振动峰, 高频区的728.91 cm-1振动峰, 为六元环上Li— F— Li的反伸缩振动。 两个位于184.32和641.34 cm-1处的弱峰, 分别归属于Li— F— Li的剪切振动和对称伸缩振动。 同时, Na原子的吸附明显提高了红外波谱的振动强度, 并随着碱金属原子的不同, 其振动特征峰有了显著的改变。 (Li2F)2K: 团簇(Li2F)2K振动峰比较复杂。 668.29 cm-1处为最强的振动峰, 为Li— F— Li三个原子的伸缩振动。 然而, 次振动峰主要分布在0~200 cm-1低频区, 主要归属于团簇骨架的伸缩振动。 与其他两个团簇相比, 此团簇的振动强度最弱, 其偶极矩为2.65 D。
采用B3LYP/6-311+G* 方法对(Li2F)nM (M=Li, Na, K; n=1, 2)团簇的几何结构、 稳定性、 电子性质和红外光谱做了理论研究。 结果发现: (Li2F)M团簇具有相似的双三角基态结构, 但是(Li2F)2M的基态结构则完全不同(例如, “ 船型” 和“ 金字塔型” )。 当n=1时, 掺杂不同碱金属原子的键能顺序为(Li2F)Li> (Li2F)Na> (Li2F)K; 但是当n=2时, 则(Li2F)2Na的键能最大。 HOMO-LOMO能隙分析得出团簇(Li2F)Li和(Li2F)2Na具有较大的能隙值, 因此这两个团簇的动力学稳定性较强。 从轨道分析发现, 团簇(Li2F)Li的HOMO和LUMO轨道都来自于Li原子的sp杂化而形成的σ 键。 然而, 团簇(Li2F)2Na的HOMO和LUMO轨道也是由sp杂化而来, 但是它们主要归咎于Li和Na两个原子的贡献。 对这些团簇电离势的分析, 发现团簇(Li2F)2K具有超低的电离势(4.23 eV), 可作为新型超碱化合物用于非线性光学。 此外, 也对这些团簇的红外光谱进行了归属; 发现碱金属(Li, Na, K)吸附在Li2F团簇表面时, 其最强红外吸收峰发生明显红移, 而碱金属(Li, K)吸附在(Li2F)2团簇时, 其红外光谱较为复杂, 主要归因于其几何结构特征。 这些发现对此类团簇材料的研究和应用提供了重要的理论基础。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|