引用本文
Ça$\dot{g}$rı Çırak, Nurettin Körözlü, Yusuf Sert, Fatih Ucun. Computational Studies on the Ground State Tautomer, Hydrogen Conformations and Vibrational Spectroscopic Analysis of Antitumor Agents: 3-Deazauracil and 6-Azauracil[J]. Spectroscopy and Spectral Analysis, 2018,38(4): 1276-1282.
Doi:10.3964/j.issn.1000-0593(2018)04-1276-07
Permissions
Doi:10.3964/j.issn.1000-0593(2018)04-1276-07
Copyright©2018, 《光谱学与光谱分析》期刊社
《光谱学与光谱分析》期刊社 所有
Computational Studies on the Ground State Tautomer, Hydrogen Conformations and Vibrational Spectroscopic Analysis of Antitumor Agents: 3-Deazauracil and 6-Azauracil
Abstract
The ground state hydrogen conformations and vibrational analysis of 3-deazauracil (3DAU) and 6-azauracil (6AU) tautomers (4-enol and 2,4-diol forms) have been calculated using ab initio Hartree-Fock (HF) and density functional theory (B3LYP) methods with 6-311++G(d,p) basis set level. The calculations have shown that the most probably preferential tautomer of 3DAU and 6AU are the 4-enol form, which gives best fit to the corresponding experimental data. The ground state conformer of the 2,4-diol form has two O—H bonds which are oriented externally and internally (to the N—H bond). The vibrational analyses of the ground state conformer of each tautomeric form of 3DAU and 6AU were done and their optimized geometry parameters (bond lengths and bond angles) were given. Furthermore, from the correlations values it was concluded that the B3LYP method is superior to the HF method for both the vibrational frequencies and the geometric parameters.
Keyword:
3-Deazauracil; 6-Azauracil; Tautomerism; Conformation; Vibrational spectra

Introduction
3DAU (4-hydroxy-2-pyridone, 2, 4-dihydroxypyridine) and 6AU (1, 2, 4-triazine-3, 5(2H, 4H)-dione) are pyridine derivative bio-molecule, having a chemical formula of [C5H5NO2] and [C3H3N3O2], respectively. These molecules are a modified nucleic acid base and, are a deaza and aza analogs of Uracil. 3DAU and 6AU have been shown antitumor activity and widely used clinically in cancer treatment. So, these pyridine derivate bio-molecules have received much attention in cancer and drug researches[1, 2, 3, 4, 5, 6]. 3DAU and 6AU have two tautomeric forms that named as 4-enol and 2, 4-diol. Each tautomeric form has different hydrogen conformations. 3DAU has 2 conformers for the 4-enol form and 4 conformers for 2, 4-diol form. 6AU has 1 conformer for 4-enol form and 4 conformers for 2, 4-diol form. So, there are many studies experimentally to determination the crystal structure and the orientation of the O— H bonds of the tautomers of 3DAU[7, 8, 9, 10, 11] and 6AU[9, 12, 13, 14].
In this present study the optimized molecular geometries and vibrational spectra of the two tautomers of all title molecules have been calculated to determine its most preferential tautomer with a established hydrogen conformation of the OH groups by using Hartree-Fock (HF) and density functional theory (B3LYP) methods. The calculated optimized geometric parameters and vibrational frequencies have been compared with the corresponding experimental data.
1 Computational Methods
The vibrational frequencies and optimized structure parameters of 3DAU and 6AU’ s tautomers have been calculated by using ab initio Hartree-Fock (HF) and density function theory (B3LYP) methods at 6-311++G(d, p) basis set level. All computations have been performed using Gauss-View molecular visualization program[15] and Gaussian 03 program package on personal computer[16]. To close the calculated frequencies to the experimental ones the scale factors of 0.905 1 and 0.961 4 are used for HF and B3LYP with 6-311++G(d, p) basis set, respectively[17]. Additionally, the calculated vibrational frequencies were clarified by means of the potential energy distribution (PED) analysis of all the fundamental vibrations modes by using VEDA 4 program[18]. VEDA 4 program have been used in previous studies by many researchers for PED analysis of vibrational modes[19, 20, 21].
2 Results and Discussion
To our knowledge, there are no theoretical studies on the vibrational assignments of 3DAU and 6AU in the literature. So, in order to introduce detailed vibrational assignments, we have performed computational analysis done by using PED analysis and the visualization of modes.
3DAU is a molecule having 13 atoms, which belongs to the point group CS. The three Cartesian displacements of the 13 atoms provide 39 internal modes, namely;
From the character table for the CS point group, since Γ trans.=2A'+A″ and Γ rot.=A'+2A″, we get
normal modes of vibration. All the vibrations are active both in infrared (IR) and Raman (R). For an N-atomic molecule, 2N-3 of all vibrations is in plane and N-3 out of plane[22]. So, for the title molecule, 6 of all the 23 vibrations are in plane and 10 are out of plane. Since the molecule is planer all the vibrations being anti-symmetric through the mirror plane of symmetry σ h will belong to the species and A″ the others being symmetric through σ h to the species A'. Thus, all the A' vibrations of thespecies will be in plane and those of the A″ species are out of plane. For 6AU, numbers of vibration modes is as follow
From the character table for the CS point group, since Γ trans.=2A'+A″ and Γ rot.=A'+2A″, we get
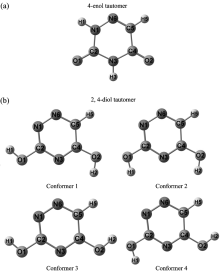
The calculated optimized possible hydrogen conformations of the tautomers of all title molecules are given in Figure 1-2. Furthermore, the total energies (corrected by zero point energy), relative energies, vibrational deviations of all the conformations of the tautomers of all title molecules have summarized in Table 1-2. The relative energy values in the tables are respect to the conformer with minimum energy. As seen from Table 1-2, the relative energies between the conformers with maximum and minimum energy are 2.00 kcal· mol-1 (HF) and 1.43 kcal· mol-1 (B3LYP) for the 4-enol form and 6.23 kcal/mol-1 (HF) and 5.92 kcal· mol-1 (B3LYP) for the 2, 4-diol form of 3DAU and 6.73 kcal· mol-1 (HF) and 6.03 kcal· mol-1 (B3LYP) for 2, 4-diol form of 6AU. As seen from the tables the conformer 1 of each tautomeric form of all title molecules has minimum energy, since, we say that the conformer 1’ s are the most stable conformers of the tautomers. So, we only take into account the conformer 1 of the two tautomers of the all title molecules in the study.
 | Fig.1 Calculated optimized structures of possible conformers of 3DAU (a) 4-enol tautomer and (b) 2, 4-diol tautomer |
{kind=link}
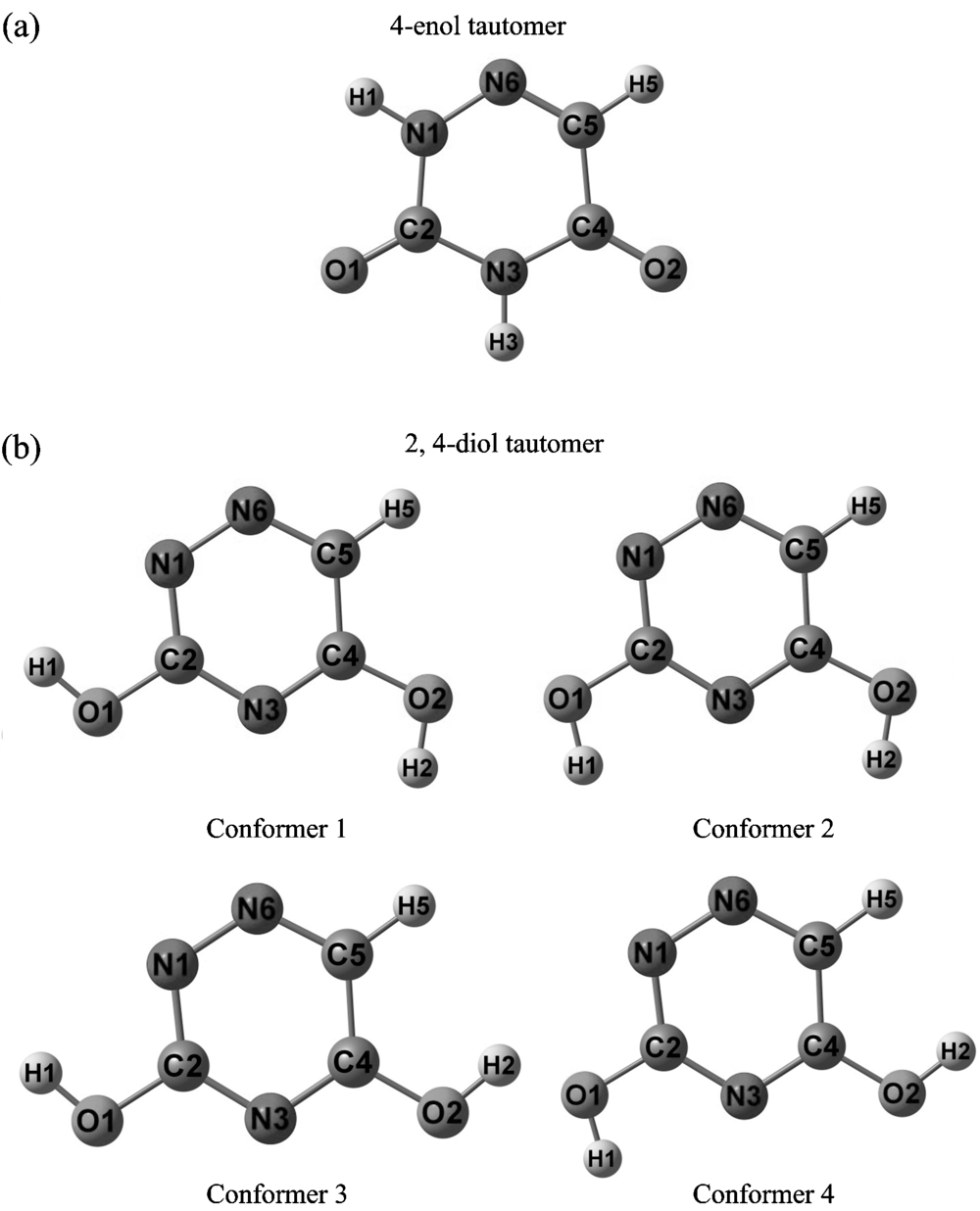 | Fig.2 Calculated optimized structures of possible conformers of 6AU (a) 4-enol tautomer and (b) 2, 4-diol tautomer |
{kind=link}
| Table 1 Total energies, relative energies and vibrational deviations for optimized conformations of 4-enol and 2, 4-diol tautomers of 3DAU |
| Table 2 Total energies, relative energies and vibrational deviations for optimized conformations of 4-enol and 2, 4-diol tautomers of 6AU |
If the total energies of the two tautomeric forms of 3DAU and 6AU in Table 1-2 are compared we can see the ground state conformer of the 4-enol form of the molecules are more stable than that of the 2, 4-diol form, with a relative energy 0.06 kcal· mol-1 (HF) and 1.80 kcal· mol-1 (B3LYP) for 3DAU and 21.00 kcal· mol-1 (HF) and 20.67 (B3LYP) for 6AU. This situation shows 4-enol tautomer of 3DAU and 6AU is more stable than 2, 4-diol tautomer. The mean vibrational deviations (|Δ ν |ave) in Table 1-2 are between the calculated vibrational frequency values of the ground state conformer and the mentioned conformer. As seen the mean vibrational deviation increases while the relative energy increases. Therefore, we state that the more different the molecular structure of the conformers is the higher the relative energy is between them, and thus, a bigger mean vibrational deviation[23, 24].
The resulting vibrationam frequencies and proposed vibrational assignments with PED’ s analysis of the ground state conformers (conformers 1) of the 4-enol and 2, 4-diol tautomers of the title molecule are given in Table 3-4, respectively. The tables also show the experimental vibrational frequencies of the compounds. The experimental (FT-IR or FT-R) vibrational frequency values have been found from the spectra obtained by the web pages of SDBS (National Institute of Advanced Industrial Science and Technology)[25]. The calculated vibrational frequencies in the tables are scaled and their symmetry species are written in the first column of the tables. As we said before all the vibrations of thespecies are in plane and those of!the species are out of plane. This was corrected by the visual inspection of all the vibrations on the Gauss-Wiev Program. As seen from Table 3-4, the calculated frequencies are in good agreement with the experimental ones. Additionally, from the correlation values in the last lines of the tables it has been clearly seen that the B3LYP method gives better correlation values than the HF method for the vibrational frequencies. The theoretical infrared intensities and Raman activities can also be seen in the tables.
| Table 3 Experimental and calculated vibrational frequencies of the ground state conformer of 4-enol tautomer of 3DAU |
| Table 4 Experimental and calculated vibrational frequencies of the ground state conformer of 4-enol tautomer of 6AU |
The calculated N— H stretching modes are in nearly 3 500 cm-1 for the 4-enol tautomer of al title molecules. These
N— H stretching modes are observed in lover frequencies due to intermolecular hydrogen bonding in FTIR spectra. In plane and out of plane bending modes of N— H bonds are in good agreement with observed spectra. One of the most important mode observed in FTIR and FT-R spectra is O=C stretching mode in 1 655 cm-1 for 3DAU and 1 755, 1 711 cm-1 for 6AU. These modes are calculated in 1 675 cm-1 for 3DAU and 1 742, 1 710 cm-1 for 6AU. The calculations approach observed data quite well. This O=C modes indicate presence of 4-enol tautomer. Additionally, O— H stretching modes are not seen in experimental spectra for 6AU. All the vibrational assignments are in excellent agreement with our recent study on 5-Bromo-2'-deoxyuridine and related biomolecules in the literature[26, 27, 28, 29].
In Table 5-6 are given the calculated optimized structure parameters (bond lengths and bond angles) for the ground state conformers of both the two tautomers of the title molecules in accordance with the atom numbers in Figure 1-2. The table also compares the calculated structure parameters with those obtained experimentally from X-ray data for the geometric structure of 3DAU[11] and 6AU[12]. The correlation values between the experimental and calculated geometric parameters can be seen in the last line of the table. From these values we can say that there is also a good agreement between them, and furthermore, the B3LYP method is superior to the HF method as it was in the vibrational frequencies.
| Table 5 Experimental and calculated geometric parameters of the ground state conformers of two tautomer of 3DAU |
| Table 6 Experimental and calculated geometric parameters of the ground state conformers of two tautomer of 6AU |
From the discussion above we can say that the ground state conformer of the 4-enol tautomer is a lower energy than that of the 2, 4-diol tautomer. So, the most probably preferential tautomer of 3DAU and 6AU are the 4-enol form. The ground state conformer of the 2, 4-diol form has two O— H bonds which are oriented externally and the internally (to the N— H bond).
3 Conclusion
The ground state hydrogen conformations and vibrational analysis of 3DAU and 6AU tautomers (4-enol and 2, 4-diol forms) have been calculated by using ab initio Hartree-Fock (HF) and density functional theory (B3LYP) methods with 6-311++G(d, p) basis set level. The calculations have shown that the most probably preferential tautomer of 3DAU and 6AU are the 4-enol form. The vibrational frequencies and optimized geometry parameters (bond lengths and bond angles) of the ground state conformer of each tautomeric form of all title molecules were obtained and compared to the corresponding experimental data. It was also seen that the B3LYP method is superior to the HF method for both the vibrational frequencies and geometric parameters.
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|