
{kind=link}
{kind=link}
{kind=link}
CO分子振转光谱的理论研究
[张学富1 , 吕兵1 , 宋晓书1, *  , 令狐荣锋
, 令狐荣锋2 ]
, 令狐荣锋|
|


作者简介: 张学富, 1989年生, 贵州师范大学物理与电子科学学院研究生 e-mail: 1208031868@qq.com
采用从头算的多参考组态相互作用(MRCI)方法并结合基组aug-cc-pCVQZ计算了CO分子基态( X1 Σ+)的势能曲线和偶极矩曲线, 得到的势能曲线、 偶极矩曲线分别与RKR势、 文献的偶极矩曲线吻合较好。 利用所得的势能, 求解双原子分子核运动的Schrödinger方程找到了CO分子 X1 Σ+态转动量子数 J=0时的70个振动态, 对于每一振动态, 分别计算了其振动能级 G( v)、 转动惯性常数 Bv和离心畸变常数 Dv, 并把计算结果与已知的42个实验值做了详细比较, 结果表明, 计算的振动能级 G( v)、 转动惯性常数 Bv、 离心畸变常数 Dv与实验值符合较好。 利用 G( v), Bv导出的光谱常数[谐振频率 ωe(2 160.1 cm-1)、 非谐振频率 ωe χe(13.1 cm-1)、 转动常数 Be(1.918 cm-1)、 振转耦合常数 αe(0.017 3 cm-1)]也与实验值的光谱常数[ ωe(2 169.8 cm-1), ωe χe(13.3 cm-1), Be(1.931 cm-1), αe(0.017 5 cm-1)]较为符合, 这在一定程度上证明了方法MRCI/Aug-cc-pCVQZ对CO分子基态性质的计算是合适而可靠的。 利用乘积近似方法计算了CO分子在常温、 中温、 高温时的配分函数, 在此基础上, 计算了CO分子在 T=296 K时的1-0跃迁带的谱线强度, 通过比较发现, 计算所得的线强度与HITRAN数据库符合较好。 进一步计算的CO分子 X1 Σ+态1-0, 2-0, 3-0, 4-0, 2-1, 3-1和4-1跃迁带的带强度也与实验值较为吻合, 同时首次计算了CO分子 X1 Σ+态3-2跃迁带、 4-2跃迁带的线强度及带强度。
The potential energy curve (PEC) and dipole moment curve (DMC) for the ground state ( X1 Σ+) of CO molecule have been computed using the multi-reference configuration interaction (MRCI) method with aug-cc-pCVQZ basis sets. Results showed that the calculated PEC, DMC are in accord with RKR, reference, respectively. With the potential energy obtained at the MRCI/aug-cc-pCVQZ level of theory, 70 vibrational states ( J=0) of the ground state of CO molecule are obtained by numerically solving the radical Schrödinger equation of nuclear motion. For each vibrational state, the vibrational energy levels G( v), the inertial rotation constants Bv and the centrifugal distortion constants Dv are reported, which accord well with the experimental values. The inertial rotation constants Bv, vibrational energy levels G( v) were fitted to determine spectroscopy constants, which the rotation coupling constant ωe(2 160.1 cm-1), the anharmonic constant ωe χe(13.3 cm-1), the equilibrium rotation constant Be(1.931 cm-1) and the vibration-rotation coupling constant αe(0.017 5 cm-1) are in good agreement with the experiment data [ ωe(2 169.8 cm-1), ωe χe(13.3 cm-1), Be(1.931 cm-1), αe(0.017 5 cm-1)], it is evident that MRCI/Aug-cc-pCVQZ is reliable for the calculation for the ground state of CO molecule. The line intensity of 1-0 transition band for the ground state of CO molecule is calculated by directly calculating the partition function at 296 K, the agreement between the calculated line intensity data and the data in HITRAN database is fairly good at 296 K. Band intensities of 1-0, 2-0, 3-0, 4-0, 2-1, 3-1, 4-1 bands are calculated for the ground state of CO molecule, which are in better agreement with the experimental values. Therefore, the line intensities and band intensities of 3-2 transition band, 4-2 transition band are firstly calculated.
引 言
CO分子是地球大气中的一种主要气体分子, 是构成大气的主要成分, 它在各种气体的电离过程中起到很大的作用, 同时它也是原子与分子物理学研究的焦点之一。 分子光谱是研究分子结构的重要手段, 其运用极其广泛, 它不仅在高分子材料、 医学、 环境检测与分析等热门领域有着重要运用[1], 而且在天体物理的研究领域也有着极其重要的地位, 特别是在研究星际大气结构上和分子云的热动力学上都有着极其重大的意义[2]。 近年来, 国内外研究CO分子的学者越来越多。 在光谱方面, 早在1969年, Toth等[3]就研究了CO分子3-0带的线强度并给出了偶极矩矩阵元, 2010年张俊丽[4]从实验的角度出发, 运用光外差-磁旋转-浓度调制(OH-MR-CMS)吸收光谱技术, 测量了CO分子在三重带d3Δ -α 3П 的吸收谱; 在获取CO分子X1Σ +态势能函数方面, Bachrach等[5]采用ACCD方法计算了CO分子的势能曲线, 并得到了谐振频率ω e与非谐振频率ω eχ e, 之后, Kobust等[6]用Hartree-Fock方法研究得到了CO分子X1Σ +态的势能曲线并导出了分子的力常数, Copper等[7]用MCSCF方法计算了CO分子的势能函数、 谐振频率ω e与非谐振频率ω eχ e。 由于受到实验条件及技术的限制, 众多文献、 实验及HITRAN数据库[8]对其X1Σ +态的3-2跃迁带光谱和4-2跃迁带光谱至今仍未见报道。 本文在高精度从头算基础上, 采用MRCI/aug-cc-pCVQZ方法对CO分子X1Σ +态计算了其势能曲线和偶极矩曲线, 发现计算得到的势能曲线与RKR[9, 10, 11, 12]势符合较好, 利用计算获得的势能求解Schrö dinger方程, 得到了CO分子X1Σ +态J=0时的70个振动能级和分子常数, 并将计算得到的振动能级和分子常数与目前已知的实验数据进行了对比。 此外, 本文利用计算得到的CO分子的配分函数, 计算了该分子在T=296 K时的1-0跃迁带的线强度, 发现计算结果与HITRAN数据库较为吻合, 在此基础上, 首次给出了CO分子X1Σ +态的3-2跃迁带、 4-2跃迁带的线强度及其带强度, 为CO分子的进一步实验和理论研究提供了参考。
在分子势能函数的计算中, 单参考组态方法、 完全组态方法在计算精度和计算量上, 很难达到令人满意的结果, 而多参考组态相互作用方法(MRCI)充分考虑了电子的动力学相关, 能够很好的自动处理对相关能有重要贡献的组态波函数(CSF), 它不仅能弥补单参考组态方法、 完全组态方法的缺陷, 而且还可以解决某些区域不收敛的问题, 被认为是当前处理相关能最好的从头算方法[13]。 CO分子共有14个电子, X1Σ +态的电子组态为1σ 22σ 23σ 24σ 21
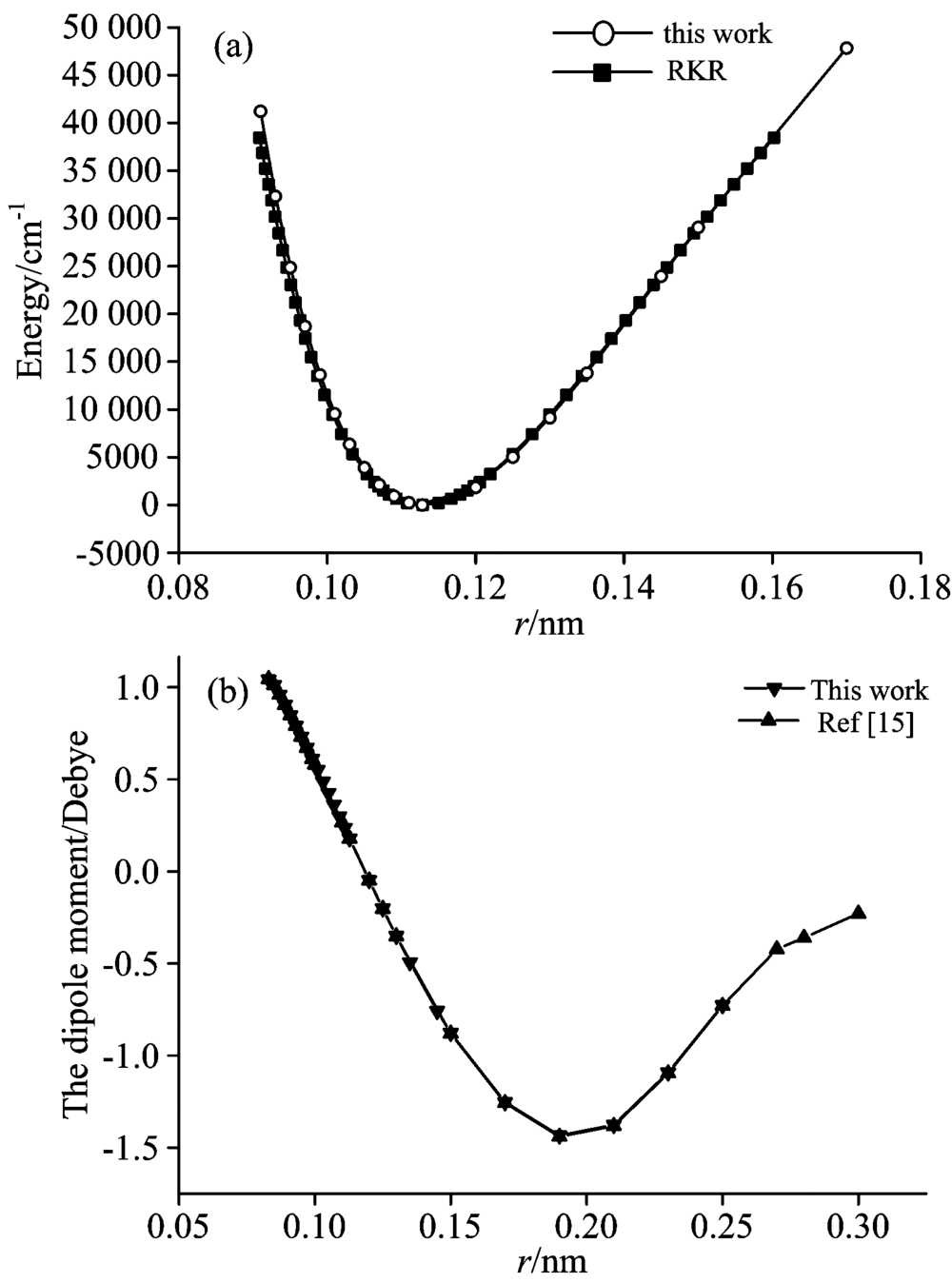 | 图1 CO分子势能曲线与偶极矩曲线 (a): 势能曲线; (b): 偶极矩曲线Fig.1 The potential energy curve and dipole moment curve of CO molecule (a): The potential energy curve; (b): The dipole moment curve |
从图1中的(a)图对比结果可知, 除了在约0.09~0.095 nm的核间距范围内, 本文计算的势能曲线与用Dunham系数拟合得到RKR势有轻微偏差外, 其余部分与RKR势基本保持一致, Dunham系数取自Chandra等[15]的研究成果。
将计算获得的势能代入分子核运动的Schrö dinger方程, 即
求解该方程找到了CO分子X1Σ +态J=0时的70个振动态, 鉴于篇幅有限, 表1只给出了CO分子的部分振动能谱, 而且得到的数值和已知的42个实验值吻合较好[16], 同时本文通过计算还产生了高于42个实验值之上的28个振动能级, 也比文献[14]多计算了64个振动能级, 虽然文献[14]得到的结果与实验值的误差比本文略小, 但文献[14]只得到了较低的6个振动态。 本文还计算得到了CO分子的转动惯性常数Bv和离心畸变常数Dv, 发现它们也与实验值较为吻合。
| 表1 CO分子常数与振动能级(J=0) Table 1 The molecular constants and vibrational levelsof CO molecule (J=0) |
采用式(2)和式(3)拟合振动能级和转动惯性常数, 得到了CO分子X1Σ +态的光谱常数, 并与实验值及其他理论结果一起列于表2。 通过比较发现, 计算的光谱常数整体上比文献更加接近实验值, 这表明本文的计算方法更适合CO分子光谱性质的计算, 能够获得更为准确的光谱常数。
| 表2 CO分子基态光谱常数 Table 2 The spectrum constants of ground state of CO |
配分函数的计算有很多方法[21], 采用了广泛使用的乘积近似方程[22], 即分子的总配分函数由两部分组成, 一是分子的转动配分函数Qrot, 二是分子的振动配分函数Qvib, 即
其中, 转动配分函数Qrot采用McDOWELL[23]的结果, 即
在式(5)中, σ 是分子的点群对称数, I是核自旋多重度, β =hc/kBT, fc是离心扭曲修正因子; 式(6)中, d=D/B, h'=H/B, 这里B, D和H是分子的转动常数, T(K)是温度, kB是波尔茨曼常数, h为普朗克常量, c为真空中的光速。 对于CO分子X1Σ +态, σ =I=1, k=0, H=0, 其转动常数B, D取自文献[20]。
对于双原子分子的振动配分函数Qvib, 采用的是谐振子近似[24], 即
在式(7)中, ν i是分子的第i个振动基频。 通过以上分析, 本文计算了CO分子的配分函数, 如表3所示。
| 表3 CO分子配分函数 Table 3 The partition function of CO molecule |
对于双原子分子, 其振转光谱从一个态|ν 'J'> 到另一个态|ν ″J″> 的线强度计算可采用式(8)
其中, Iif, ν if, Ei, T和Aif分别是线强度[单位: cm-1· (molecule· cm-2)-1]、 跃迁波数(单位: cm-1)、 低态能量(单位: cm-1)、 温度(单位: K)、 爱因斯坦系数(单位: s), Q是配分函数, J'是跃迁上态的转动量子数, 其中, J'=J-1, 对应跃迁带的P支谱线, J'=J+1, 对应跃迁带的R支谱线, J'=J, 则对应跃迁带的Q支谱线。 公式中的ν if, Aif等可通过Level 8.0[25]计算得到。 所有线强度的总和为带强度, 即
式(9)中, Isum是带强度[单位: cm-1· (molecule· cm-2)-1]。
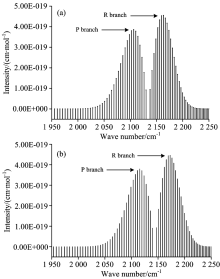
综合以上的分析, 本文研究了CO分子X1Σ +态在T=296 K时1-0跃迁带的光谱, 计算了跃迁波数在1 955.606~2 256.675 cm-1范围内的80条谱线, 其中, R支谱线40条, P支谱线40条, 并将之与HITRAN数据库做了比较, 如图2所示。
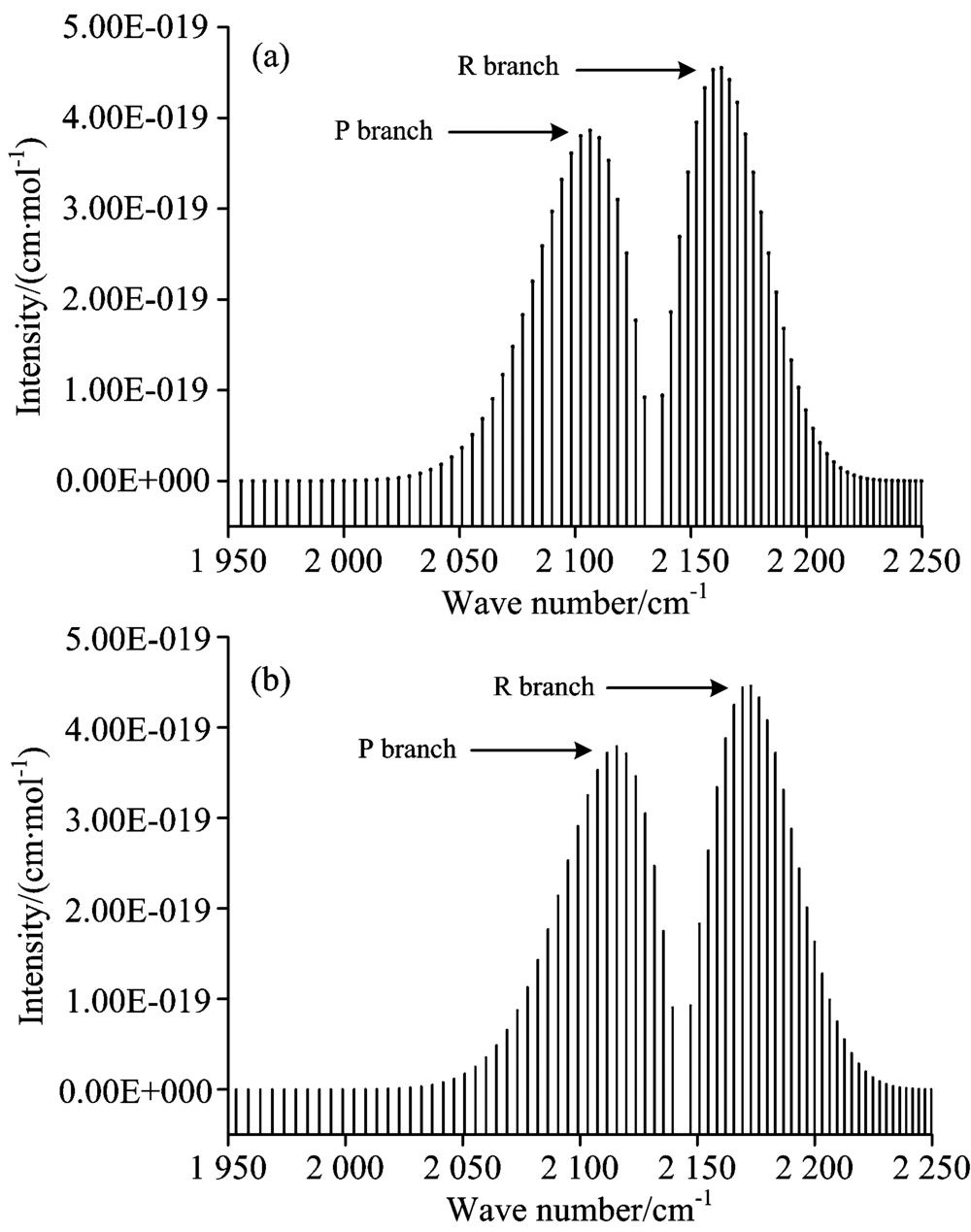 | 图2 CO分子在296 K时模拟光谱与HITRAN数据库对比 (a): 296 K, 本文计算; (b): 296 K, HITRAN数据库Fig.2 Comparison of calculated simulated spectra in the 1-0 with those in HITRAN database for CO at 296 K (a): 296 K, calculated in this work; (b): 296 K, extrapolated from HITRAN database |
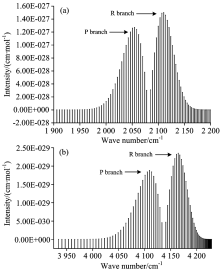
基于计算得到的分子势能与RKR势略有偏差, 以致于计算得到的跃迁波数比实验值略低了约15 cm-1, 这使计算得到的P支谱线、 R支谱线的跃迁波数均相对于实验值略小, 导致图1中的(a)图谱线相对于(b)图谱线均轻微向左偏移约15 cm-1。 通过比较发现, 本文计算的最大线强度[4.55× 10-19 cm-1· (molecule· cm-2)-1]出现在J=7的R支谱线, 它与实验值[4.46× 10-19 cm-1· (molecule· cm-2)-1]符合较好, 计算的最小线强度[9.19× 10-25 cm-1· (molecule· cm-2)-1]出现在J=40的P支谱线, 也与实验值[8.15× 10-25 cm-1· (molecule· cm-2)-1]较为吻合, 而且P支谱线强度整体上略小于R支谱线强度。 在此基础上, 本文进一步的研究了CO分子X1Σ +态在T=296 K时的3-2跃迁带、 4-2跃迁带的光谱特性, 对3-2跃迁带, 计算了跃迁波数在1 906.051 6~2 201.579 cm-1范围内的跃迁谱线强度, 其中P支谱线40条, R支谱线40条, 最大的线强度位于J=8的R支谱线, 其值为1.4496× 10-27 cm-1· (molecule· cm-2)-1; 对于4-2跃迁带, 本文也计算了跃迁波数在3 934.514 2~4 228.657 6 cm-1范围内的80条跃迁谱线强度, 最大线强度出现在J=8的R支谱线, 其值为2.347 3× 10-29 cm-1· (molecule· cm-2)-1。 图3给出了3-2跃迁带、 4-2跃迁带的模拟光谱。
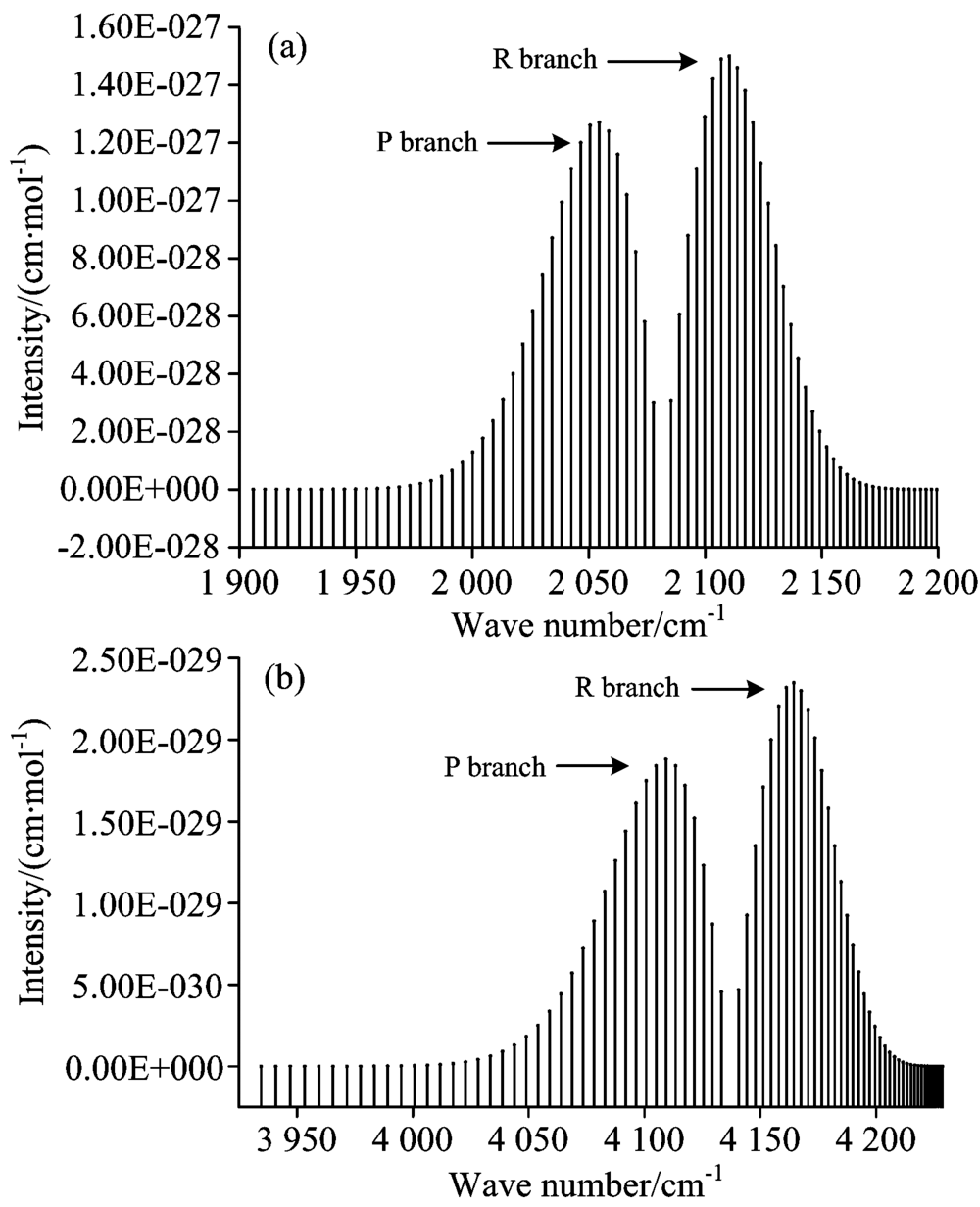 | 图3 CO分子在296K时的模拟光谱 (a): 3-2带光谱; (b): 4-2带光谱Fig.3 Simulated spectra of CO molecule at 296 K (a): Spectrum of 3-2 transition band; (b): Spectrum of 4-2 transition band |
从图2、 图3可以看出, 计算得到的所有跃迁谱线中没有Q支谱线产生, 这是因为CO分子基态之间的跃迁属于1Σ -1Σ 跃迁, 根据选择定则, 对于1Σ -1Σ 态之间的跃迁, ∇J=0的跃迁是禁止的, 所以这类谱带结构中没有Q支谱线。
对于CO分子X1Σ +态, 进一步计算了带强度, 表4列出了计算值与HITRAN数据库的实验值, 从表4中不难发现, 计算1-0, 2-0, 3-0, 4-0, 2-1, 3-1, 4-1带的带强度和HITRAN数据库的结果符合较好。 同时, 也给出了CO分子基态3-2跃迁带和4-2跃迁带的带强度, 如表4所示。
| 表4 CO分子基态跃迁带强度[unit: cm-1· (molecule· cm-2)-1] Table 4 Band intensity of ground state of CO molecule [unit: cm-1· (molecule· cm-2)-1] |
利用高精度的从头算方法MRCI/aug-cc-pCVQZ计算了CO分子基态(X1Σ +)的势能曲线和偶极矩曲线, 利用所得的势能函数, 通过求解核运动的Schrö dinger方程, 找到了CO分子X1Σ +态J=0时的70个振动态, 并求出了每个振动态的振动能级G(ν )、 转动惯性常数Bv和离心畸变常数Dv, 计算结果与已知的42个实验值符合较好, 进一步导出的光谱常数也与实验值较为符合。 其次, 采用乘积近似方法计算了CO分子的配分函数, 其中, 振动配分函数采用了谐振子模型, 转动配分函数则采用了非刚性转子模型, 并且考虑了离心扭曲修正。 在此基础上, 计算了CO分子X1Σ +态的1-0跃迁带的线强度, 计算结果与HITRAN数据库的实验值较为吻合, 同时计算的CO分子基态的1-0, 2-0, 3-0, 4-0, 2-1, 3-1和4-1带的带强度也与HITRAN数据库符合较好。 本文计算获得了CO分子X1Σ +态的3-2跃迁带、 4-2跃迁带的线强度及带强度, 虽然目前尚没有实验数据和理论计算结果与之对比, 但希望今后能有更多的研究工作为其报道, 以促进对CO分子性质的全面认识。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|