
{kind=link}
{kind=link}
{kind=link}
Para-Xylene分子结构和光谱的外场效应研究
[杜建宾1, 2, *  , 张倩
, 张倩1 , 李奇峰2 , 唐延林3 ]
, 张倩|
|


作者简介: 杜建宾, 1979年生, 廊坊师范学院讲师, 天津大学精密仪器与光电子工程学院博士研究生 e-mail: dujianbinfzf@sina.com
对二甲苯(PX)是一种重要的化工原料, 广泛地用于合成聚脂纤维树脂, 是医药和农药用聚合单体, 因此了解对二甲苯分子结构和光谱的外场效应具有十分重要的意义。 为研究外电场对PX分子结构和红外(IR)光谱产生的影响, 本文采用密度泛函理论(DFT)B3LYP方法在6-311++G(d, p)基组水平上优化了不同外电场(0~0.030 a.u., 0~1.542×1010 V·m-1)作用下PX分子的基态几何结构, 在此基础上利用同样的方法计算了PX分子在不同电场下的振动频率, 得到了分子的IR光谱, 最后对PX的分子结构和IR光谱受外电场作用的的影响规律进行了研究。 结果表明: 频率497 cm-1的吸收峰为苯环上的C1-H7和C2-H8的面外摇摆振动, 812 cm-1的吸收峰是苯环上的C4-H9和C5-H10的面外摇摆振动, 1 547 cm-1为苯环上的C1-H7和C2-H8的面内弯曲振动, 3 017 cm-1为甲基上的C11-H14和C15-H17的伸缩振动所产生, 3 164 cm-1为苯环上的C4-H9和C5-H10的伸缩振动所产生; 外电场与分子内电场的叠加效应使得 R(4, 9), R(5, 10), R(11, 14)和 R(15, 17)等的键长随着外电场的增强急剧增长, 而 R(1, 7), R(2, 8), R(11, 12) 和 R(15, 16)等随着外电场的增强变化不明显; 随着 R(4, 9)和 R(5, 10)的增加, 吸收峰Ⅱ和Ⅴ出现了明显的红移, 它们的频率分别减少了133和140 cm-1, 峰Ⅳ在外电场较弱时频率增加, 而当外电场较强时频率又开始减小, 峰Ⅰ和Ⅲ的频率变化不明显。 总之, 在外电场的作用下, 分子结构变化剧烈, 红外光谱吸收峰出现了红移或蓝移, 伴随着吸收峰的移动, 分子的摩尔收系数ε也进行了重新分配, 分子的振动斯塔克效应(VSE)明显。
Para-xylene(PX)is an important chemical raw materials. In order to study the influence of external electrical field on molecular structure and infrared spectra of PX, the method B3LYP of the density functional theory at 6-311++G(d, p) level has been used to calculate geometrical parameters and infrared(IR) spectra under different external electric fields ( from 0 to 0.030 a. u.) in this article. The results show that the most strongest absorption of IR spectra of PX is produced by C11-H14 and C15-H17 stretching vibration; the molecular geometry parameters is strongly dependent on the external field intensity; the significant negative (“red”) and positive (“blue”) frequency shifts, the redistribution of molar absorption coefficient are observed, i. e., vibrational Stark effect(VSE) is obvious.
近年来, 随着我国聚酯行业的快速发展, 对二甲苯(para-xylene, PX)的市场需求与日俱增。 作为一种重要的基础化学品, 对二甲苯主要用于生产精对苯二甲酸, 进而生产聚酯, 还可用作溶剂以及医药、 香料、 油墨等行业的生产原料[1]。 近年来, 对有机污染物的降解和检测受到广泛重视, 人们研究了通过联合高铁酸盐和光催化氧化、 增强型电场协助光催化降解有机污染物[2], 但对于PX在外电场下的分子结构和红外(infrared, IR)光谱的理论计算目前还未见报道。 在外电场的作用下, 许多新的化学变化和现象会出现, 如新激发态出现、 化学键碎裂、 新自由基产生[3]和振动斯塔克效应(外电场改变分子结构和红外光谱的振动频率)等。 本文采用密度泛函理论(density functional theory, DFT)[4, 5, 6]方法, 研究了不同外电场对PX分子的基态几何结构和IR光谱产生的影响, 这为以PX为原料合成的有机污染物的降解和检测方法研究提供了理论依据。
在外电场作用下, 分子体系的总能量为
其中, E0是无电场时分子的总能量, Fi(i=1-3)是外电场强度, ui是分子的电偶极矩, β ijk和γ ijkl分别是一阶和二阶极化率, 在这里,
ρ (
式(3)中, α ij为线性极化率, β ijk和γ ijkl是非线性极化率。 分子振动频率的改变量Δ ω 和外电场的关系为
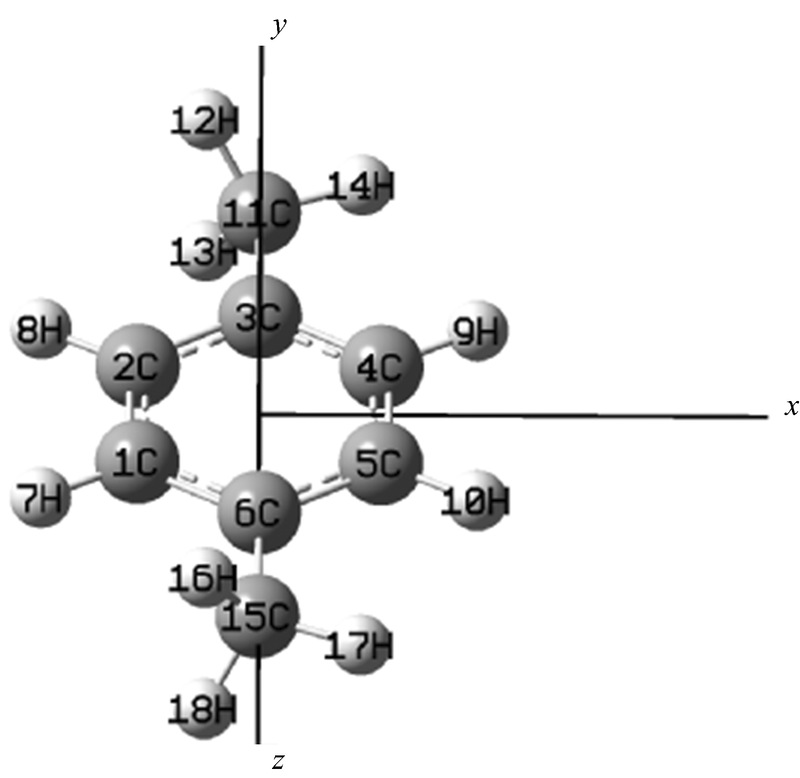 | 图1 PX分子结构示意图Fig.1 Optimized molecular structure of PX |
PX分子的结构如图1所示, 为分析外电场对PX分子的影响, 本文对PX分子沿x轴方向上加上一系列强度为0~0.030 a.u.(0~1.542× 1010 V· m-1)外电场[7], 采用DFT/B3LYP方法在6-311++G(d, p)基组水平上对其分子几何结构进行优化, 在此基础上采用同样的方法计算了分子的红外光谱。 全部计算在Gaussian09软件包中完成[8, 9]。
采用DFT优化了不同外电场(0~0.030 a.u.)作用下PX分子的基态几何结构。 随着外电场的增加, PX的几何参数对电场强度的大小有着明显的依赖关系, 如图2所示, R(4, 9), R(5, 10), R(11, 14)和R(15, 17)等的键长随着外电场的增强急剧增长, 而R(1, 7), R(2, 8), R(11, 12) 和R(15, 16)等随着外电场的增强变化不明显, 这些现象可以用外电场与分子内电场的叠加效应来定性的解释[3]。 伴随着外电场的增强, 电子的局域转移使得4C-9H, 5C-10H, 11C-14H和15C-17H之间的电场减弱, 从而键长增加: 这些C— H中C的电负性较强, 外电场的方向与其电子云的偏移方向相反, 即外电场与内电场方向相反, 所以外电场对其内电场削减较强, 也就使得这些C— H的键长增长非常明显; 与之相反, 外电场与内电场的叠加使得1C-7H和2C-8H之间的电场增强, 但由于外电场接近分子内电场的大小, 所以R(1, 7), R(2, 8)等不像R(4, 9), R(5, 10)等的变化那么明显。
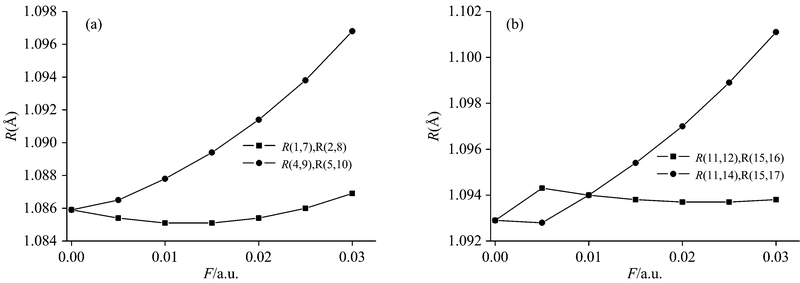 | 图2 PX分子主要键长R随外电场变化的关系Fig.2 The relation of the main bond length and electric field intensity of PX |
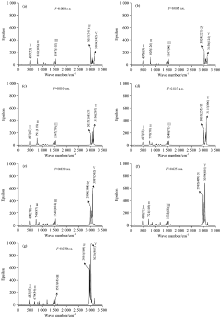
采用DFT/B3LYP方法优化了PX分子的基态几何结构, 在此基础上通过计算PX的频率得到了它的IR光谱, 如图3(a)所示, 频率497 cm-1吸收峰为苯环上的C1-H7, C2-H8的面外摇摆振动, 812 cm-1的吸收峰是苯环上的C4-H9和C5-H10的面外摇摆振动, 1 547 cm-1为苯环上的C1-H7和C2-H8的面内弯曲振动, 3 017 cm-1为甲基上的C11-H14和C15-H17的伸缩振动所产生, 3 164 cm-1为苯环上的C4-H9和C5-H10的伸缩振动所产生。 表1为理论计算和Sadtler红外数据库[10]的PX分子数据对比, 可以看出, 计算数据和实验数据符合较好, 尤其在指纹区的497和812 cm-1的两个吸收峰和实验值分别只相差13和17 cm-1, 这说明我们的计算结果是可信的。
| 表1 无外场时理论计算和Sadtler数据库的PX分子IR数据(cm-1) Table 1 The calculated and Sadtler’ s data of PX molecule without external electric fields |
 | 图3 PX分子在不同外电场作用下的IR光谱Fig.3 The infrared spectra of PX under different external electric fields |
采用DFT/B3LYP方法在6-311++G(d, p)基组水平上计算了PX分子不同外电场下的频率, 得到了它们的IR光谱, 如图3所示, 在这里我们只列出比较强的5个谱峰。 从图3中我们可以看到分子在外电场中出现了强烈的振动斯塔克效应(vibrational stark effect, VSE): 峰Ⅱ 和Ⅴ 出现了明显的红移, 它们的频率分别减少了133和140 cm-1, 这是由于吸收峰Ⅱ 和Ⅴ 分别是C4-H9和C5-H10的面外摇摆振动、 伸缩振动产生的, C4-H9和C5-H10的键长从1.085 9 Å 变化到了1.096 8 Å , 增加了0.010 9 Å (如图2所示); 峰Ⅳ 在外电场较弱时频率增加, 而当外电场较强时频率又开始减小, 这是由于C11-H14和C15-H17的键长先减小后增加造成的; 峰Ⅰ 和Ⅲ 的频率变化不明显, 也是由于C1-H7和C2-H8的键长变化不明显的原因(如图2所示)。 伴随着吸收峰的红移或蓝移, 分子的摩尔吸收系数ε 也进行了重新分配。
采用密度泛函理论计算得到了不同外电场下PX的分子结构和IR光谱, 结果表明: 分子的最强红外吸收峰为甲基上的C11-H14和C15-H17的伸缩振动所产生; 在外电场的作用下, 分子结构变化剧烈, 红外光谱吸收峰出现红移或蓝移, 分子的摩尔吸收系数重新分配, 振动斯塔克效应明显。 这些工作不仅为以PX为原料合成的有机污染物的降解和检测方法研究提供了理论依据, 也对其他环境毒物的检测方法和降解机理的研究有着启示作用。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|