引用本文
[J]. 光谱学与光谱分析, 2018,38(11): 3631-3637.
Rubarani P Gangadharan, S Sampath Krishnan. Quantum Chemical Calculations on 4-[2-(Tert-Butylamino)-1-Hydroxyethyl]-2-(Hydroxymethyl) Phenol by Density Functional Theory[J]. Spectroscopy and Spectral Analysis, 2018,38(11): 3631-3637.
Doi:10.3964/j.issn.1000-0593(2018)11-3631-07
Permissions
Rubarani P Gangadharan, S Sampath Krishnan. Quantum Chemical Calculations on 4-[2-(Tert-Butylamino)-1-Hydroxyethyl]-2-(Hydroxymethyl) Phenol by Density Functional Theory[J]. Spectroscopy and Spectral Analysis, 2018,38(11): 3631-3637.
Doi:10.3964/j.issn.1000-0593(2018)11-3631-07
Copyright©2018, 《光谱学与光谱分析》期刊社
《光谱学与光谱分析》期刊社 所有
Quantum Chemical Calculations on 4-[2-(Tert-Butylamino)-1-Hydroxyethyl]-2-(Hydroxymethyl) Phenol by Density Functional Theory

Abstract
Density functional theory (DFT) calculations have been carried out for the compound 4-[2-(tert-butylamino)-1-hydroxyethyl]-2-hydroxymethyl) phenol (4BAHEHMP) by using the B3LYP method at the 6-311++G (d,p) basis set level. The electric dipole moment ( μ) and the first hyperpolarizability (α) values of the investigated molecule were computed. Total and partial density of state (TDOS and PDOS) and also overlap population density of state (COOP or OPDOS) diagrams analysis were presented. HOMO and LUMO energies confirm that charge transfer occurs within the molecule. In addition Molecular Electrostatic Potential (MEP), Natural Bond Orbital analysis (NBO) and Non- Linear Optical (NLO) properties are studied.
Keyword:
DFT; NBO; Density of states

Introduction
4-[2-(tert-butylamino)-1-hydroxyethyl]-2-(hydroxymethyl) phenol salbutamol) is a short-acting β 2-adrenergic receptor agonist. Its molecular formula is C13H21NO3. It is used for treatment of breathing problems in patients who have asthma or chronic obstructive pulmonary disease (COPD). Usually used in the form of sulphate salt, salbutamol was developed because its molecule is similar to adrenaline, a natural bronchodilator produced by human body. During an asthma or COPD attack, the bronchioles that feed air deep into the lungs become inflamed and narrowed due to muscles in their walls contracting, which is very dangerous and can be life-threatening. In this case body’ s natural hormone, adrenaline which has a specific shape that helps the molecule fit into active sites on the cells in the muscle walls, will bring about muscles relaxation, thus cause the airways to dilate. This process is called bronchodilation. Salbutamol, along with other modern bronchodilators, was therefore developed based on the mechanism that adrenaline works on the bronchioles[1].
We have selected 4-[2-(tert-butylamino)-1-hydroxyethyl]-2-(hydroxymethyl) phenol, abbreviated as 4BAHEHMP for our study. Various properties like structure geometry, Non-Linear Optical (NLO) property, Natural Bond Orbital (NBO), Highest Occupied Molecular Orbital (HOMO), Lowest Unoccupied Molecular Orbital (LUMO) energies and Molecular Electrostatic Potential (MEP) analysis of the 4BAHEHMP are performed to elucidate the information regarding charge transfer within the molecule. To understand structure property relationship, HOMO-LUMO and natural bond orbitals have been obtained.
1 Computational Details
Computational aspects for geometry optimization and electronic structure of the compound have been done by density functional theory[2] by using the Gaussian 03W program package[3] employing different basis sets and Becke’ s three parameter hybrid exchange functionals with Lee-Yang-Parr correlation functionals (B3LYP)[4, 5, 6]. All the calculated vibrational wavenumbers reported in this study are the scaled values. In order to investigate intra-molecular charge transfer interactions, rehybridization and delocalization of electron density within the molecule, the natural bonding orbitals (NBO) analysis has been performed. The main natural orbital interactions were analyzed on the basis of NBO calculations done at DFT/B3LYP level using Gaussian 03W package. In the NBO analysis[7, 8] the electronic wave functions are interpreted in terms of a set of occupied Lewis-type (bond or lone pair) and a set of unoccupied non-Lewis (antibond or Rydberg) localized NBO orbitals. Delocalization of electron density (ED) between these orbitals corresponds to a stabilizing donor-acceptor interaction. The second-order perturbation theory has been employed to evaluate the stabilization energies of all possible interactions between donor and acceptor orbitals in the NBO basis. To calculate functional group contributions to the molecular orbitals, the total density of states (TDOS or DOS) spectrum was prepared by using the program GaussSum 2.2[9]. The contribution of a group to a molecular orbital was calculated by using Mulliken population analysis.
2 Results and Discussion
2.1 Geometry Optimization
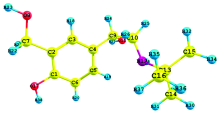
The theoretical structure of molecule have been calculated by DFT using B3LYP functional having basis set 6-31G (d, p) with the help of Gaussian 03W package and geometry obtained from B3LYP/6-311++G (d, p) is shown in Fig.1.
 | Fig.1 Optimized molecular structure and atomic numbering of 4BAHEHMP |
{kind=link}
2.2 HOMO-LUMO Analysis
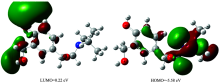
The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are the main orbitals that plays an important role in chemical stability. The HOMO exhibits the ability to donate an electron and LUMO as an electron acceptor serves the ability to obtain an electron. The HOMO and LUMO energy calculated by B3LYP/6-31++G (d, p) level of theory show the energy gap which reflects the chemical activity of the molecule.
HOMO energy (B3LYP)=-5.58 eV
LUMO energy (B3LYP)=0.22 eV
HOMO-LUMO energy gap (B3LYP)=5.80 eV
The atomic orbital compositions of the frontier molecular orbitals are shown in Fig.2.
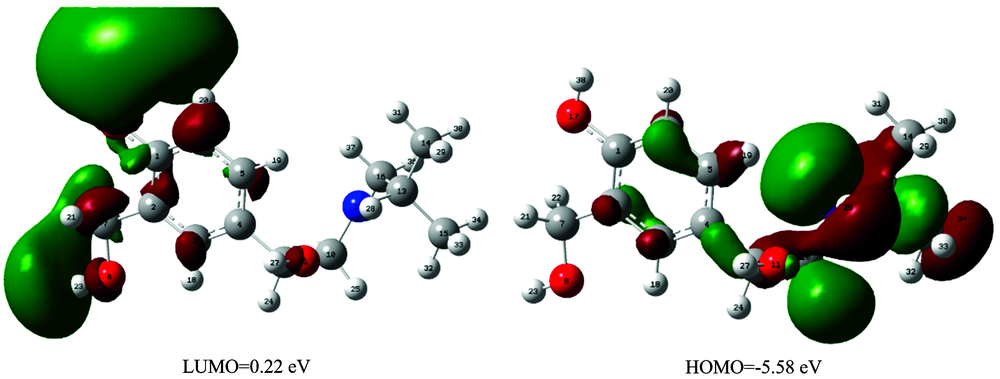 | Fig.2 The molecular orbitals of 4BAHEHMP at B3LYP/6-31++G(d, p) level |
{kind=link}
On the basis of HOMO-LUMO energies global reactivity descriptors, such as the energies of frontier molecular orbitals (EHOMO, ELUMO), energy band gap (Ε HOMO-Ε LUMO), electronegativity (χ ), chemical potential (μ ), global hardness (η ), global softness (S) and global electrophilicity index (ω ), which describe the electrophilic behaviour[10, 11, 12, 13, 14], have been calculated for 4BAHEHMP using Equations (1)— (5).
2.3 Natural Bond Orbital Analysis
NBO analysis is an efficient method for study of the intramolecular and intermolecular bonding and interactions among bonds. This analysis also provides the study of filled NBOs (donors) and empty NBOs (acceptors) and their interactions with the stabilization energy E(2) resulting from the second-order perturbation theory. The larger the E(2) value, the more intensive is the interaction between electron donors and acceptors, i.e. the more electron donating tendency from electron donors to acceptors and the greater the extent of conjugation of the whole system. This interaction results a loss of occupancy from the concentration of electron NBO of the idealized Lewis (bond or lone pair) structure into an empty (anti-bond or Rydberg) non-Lewis orbital. The second-order perturbation theory analysis of Fock matrix in NBO basis of 4BAHEHMP molecule display strong intra-molecular conjugative and hyperconjugative interactions and demystify the rehybridization and delocalization of electron density within the molecule. Some important interactions between Lewis and non-Lewis orbitals along with their interacting stabilization energies are shown in Table 1.
| Table 1 Second order perturbation theory analysis of Fock matrix in NBO basis for 4BAHEHMP |
The Fock matrix analysis shows strong intra-molecular hyperconjugative interactions of π electrons between π bond orbitals and antibonding orbitals. These interactions are established by the orbital overlapping between π (C— C or C— H) and π * (C— C or C— H) bond orbitals resulting ICT (Intramolecular charge transfer) causing stabilization of the system. The electron density (ED) at the six conjugated π bonds (1.5~1.7e) and π * antibonds (0.1~0.4 e) of the phenyl ring clearly shows strong delocalization leading to stabilization of energy in the range of 15~25 kcal· mol-1. These results are consistent with as reported by James[15]. A strong intramolecular interaction of π electrons occurs from π (C3— C4) and π (C1— C2) bonds to the π * (C5— C6) and π * (C3— C4) antibonds corresponding to the stabilization energies 23.07 and 20.96 kcal· mol-1 respectively. The most important interaction energy in this molecule is electron donating from O8 LP (2) to the antibonding acceptor σ * (C7— H22) resulting moderate stabilization energy of 6.28 kJ· mol-1. The maximum energy delocalization take part in the π — π * transition. These intramolecular charge transfer (n— σ * , n— π * and π — π * ) may induced biological activities in the molecule. The E(2) values and types of the transition is shown in Table 1.
2.4 Total, Partial, and Overlap Population Density-of-States
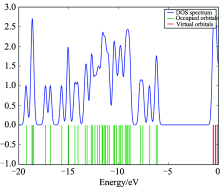
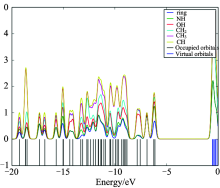
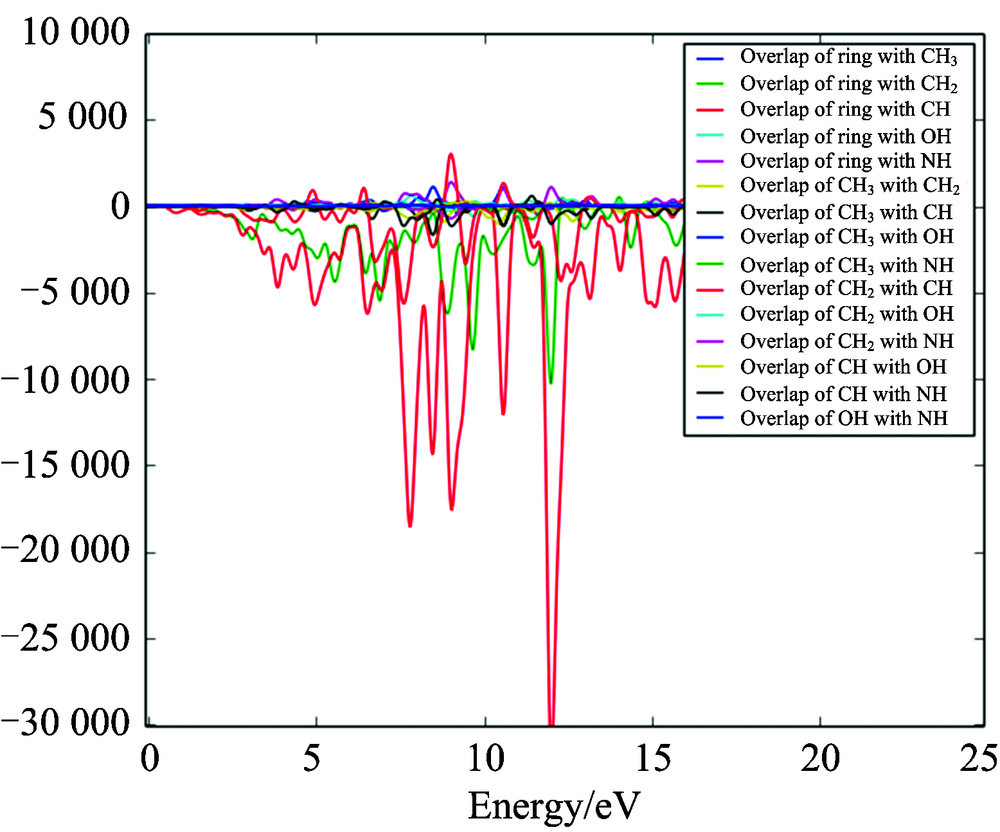
Consideration of only the HOMO and LUMO may not yield a realistic description of the frontier orbitals, because in the boundary region, neighbouring orbitals may show quasi degenerate energy levels. For this reason, the total (TDOS), partial (PDOS), and overlap population (OPDOS or COOP (Crystal Orbital Overlap Population)) density of states[16, 17, 18], in terms of Mulliken population analysis are calculated and created by convoluting the molecular orbital information with Gaussian curves of unit height and full width at half maximum (FWHM) of 0.3 eV by using the GaussSum 2.2 program. Figures 3— 5 represent the TDOS, PDOS and OPDOS plot of 4BAHEHMP, respectively. The most important application of the DOS plots is to demonstrate MO compositions and their contributions to the chemical bonding through the OPDOS plots, which are also referred in the literature as COOP diagrams.
The bonding, antibonding and nonbonding natures of the interaction of the two orbitals, atoms or groups are shown by OPDOS diagram. A positive value of the OPDOS indicates a bonding interaction (because of the positive overlap population), negative value means that there is an anti-bonding interaction (due to negative overlap population) and zero value indicates nonbonding interactions[19]. Additionally, the OPDOS diagrams allow us to determine and compare of the donor-acceptor properties of the ligands and ascertain the bonding and non-bonding. The calculated total electronic density of states (TDOSs) diagrams of the 4BAHEHMP is given in Fig.3. The partial density of state plot (PDOS) mainly presents the composition of the fragment orbitals contributing to the molecular orbitals which is seen from Fig.4. As seen Fig.4, HOMO orbitals are localized on the ring and their contributions are about 50%. The LUMO orbitals are localized on the ring (91%) of the compound. Only based on the percentage shares of atomic orbitals or molecular fragments in the molecule is difficult to compare groups in terms of its bonding and anti-bonding properties. To unravel some more detail in the bonding of the molecule, we have plotted the overlap population density of states. Fig.5 shows the OPDOS for selected groups i.e., between ring with CH3, CH2, CH, OH, CH3 with CH2, CH, OH, NH, CH2 with CH, NH, OH, CH with OH, NH and OH with NH atoms.
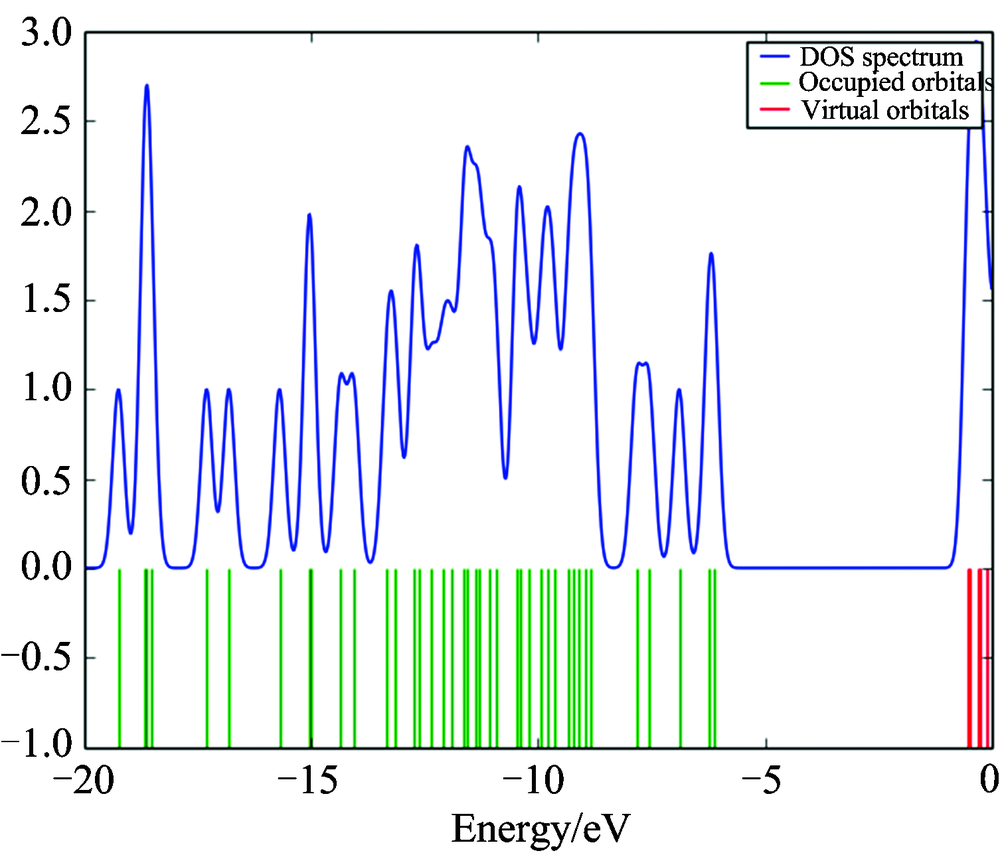 | Fig.3 The calculated TDOS diagram of 4BAHEHMP |
{kind=link}
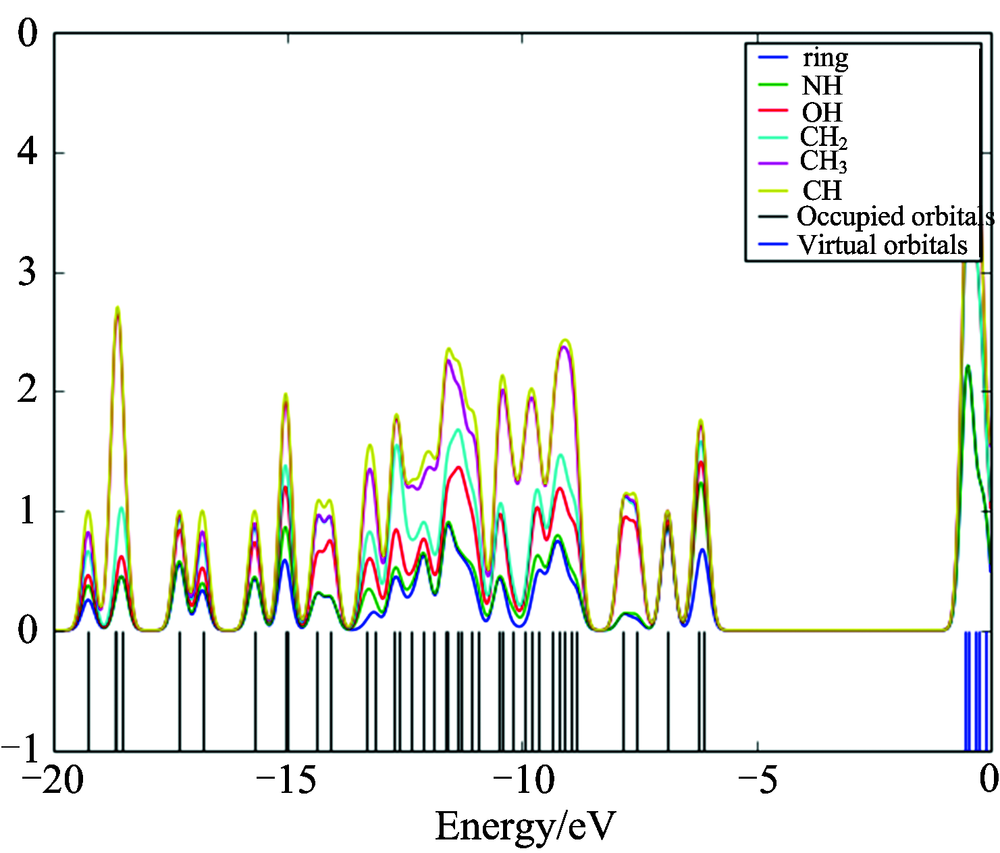 | Fig.4 The calculated PDOS diagram of 4BAHEHMP |
{kind=link}
 | Fig.5 The OPDOS (or COOP) diagram of 4BAHEHMP |
{kind=link}
Some of orbitals of energy values of interaction between selected functional groups which are shown from figures easily, (C— H) atoms overlapped by CH2 (red line) and (N— H) atoms overlapped by CH3(green line) are negative (anti-bonding interaction). As can be seen from the OPDOS plots for the 4BAHEHMP have anti-bonding character in frontier HOMO and LUMO molecular orbitals for phenyl and oxygen atoms. Thus, the OPDOS functions enable one to ascertain the bonding characteristics of electronic levels in a given energy range with respect to any pair of molecular fragments.
2.5 Molecular Electrostatic Potential
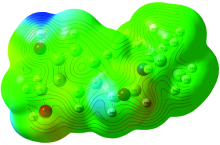
Electrostatic potential maps, are also known as electrostatic potential energy maps, or molecular electrical potential surfaces, and they illustrate the charge distributions of molecules three dimensionally. The purpose of finding the electrostatic potential is to find the reactive site of a molecule. These maps allow us to visualize variably charged regions of a molecule. Knowledge of the charge distributions can be used to determine how molecules interact with one another. Molecular electrostatic potential (MEP) mapping is very useful in the investigation of the molecular structure with its physiochemical property relationships[20, 21, 22, 23]. Total SCF electron density surface mapped with molecular electrostatic potential (MEP) of 4BAHEHMP are shown in Fig.6. The molecular electrostatic potential surface MEP which is a 3D plot of electrostatic potential mapped onto the iso-electron density surface simultaneously displays molecular shape, size and electrostatic potential values. The MEP which is a plot of electrostatic potential increases in the order red< orange< yellow< green< blue. The colour scheme for the MEP surface is red-electron rich or partially negative charge; blue-electron deficient or partially positive charge; light blue-slightly electron deficient region; yellow-slightly electron rich region, respectively. Areas of low potential, red are characterized by an abundance of electrons. Areas of high potential, blue are characterized by a relative absence of electrons. As can be seen from the MEP’ s map of the molecule the regions having the negative potential are over the oxygen atoms and nitrogen (N12). The MEP map shows that the negative potential sites are on electronegative atoms as well as the positive potential sites are around the hydrogen atoms. From these results we can say that the hydrogen atoms indicates the strongest attraction and the oxygen atom indicates the strongest repulsion.
 | Fig.6 Molecular Electrostatic Potential (MEP) Map for 4BAHEHMP |
{kind=link}
2.6 Nonlinear Optical Properties and Dipole Moment
Density functional theory has been used as an effective method to investigate the organic non-linear optical (NLO) materials. Recent research works have illustrated that the organic non-linear optical materials are having high optical non-linearity than inorganic materials[24]. In the presence of an applied electric field, the energy of a system is a function of the electric field. Polarizabilities and hyperpolarizabilities characterize the response of a system in an applied electric field[25]. They determine not only the strength of molecular interactions but also the cross sections of different scattering and collision processes, as well as the NLO properties of the system[26, 27]. For this subject, in this study the electronic dipole moment, molecular polarizability, anisotropy of polarizability and molecular first hyperpolarizability of present compound were investigated. The polarizability α and the hyperpolarizability β and the electric dipole moment μ of the compound are calculated by finite field method using B3LYP/6-311++G (d, p) basis set available in Gaussian 03 package. The polarizability and hyperpolarizability tensors (α xx, α xy, α yy, α xz, α yz, α zz and β xxx, β xxy, β xyy, β yyy, β xxz, β xyz, β yyz, β xzz, β yzz, β zzz) can be obtained by a frequency job output file of Gaussian. However α and β values of Gaussian output are in atomic units (a.u.) so they have been converted into electronic units (esu) (α ; 1 a.u.=0.148 2× 10-24 esu, β ; 1 a.u.=8.639 3× 10-33 esu). The complete equations for calculating the magnitude of total static dipole moment μ , the mean polarizability total α tot, the anisotropy of the polarizability Δ α and the mean first hyperpolarizability β tot can be calculated using the Equations (1— 5). The calculated values of dipole moment [μ (D)], polarizability (α ) and first hyperpolarizability (β ) are tabulated in Table 2. The magnitude of the molecular hyperpolarizability β , is one of key factors in NLO system. The calculated first static hyperpolarizability β tot value is equal to 3.427× 10-30 esu. Domination of particular component indicates on a substantial delocalization of charges in that direction. It is noticed
| Table 2 The units for dipole moment μ (D), polarizability (Δ α ), the average polarizability (α ) and hyperpolarizability are (a.u) (β tot) of 4BAHEHMP calculated by B3LYP method |
that in β xxx direction, the biggest values of hyperpolarizability are noticed and subsequently delocalization of electron cloud is more in that direction. The maximum β value may be due to p-electron cloud movement from donor to acceptor which makes the molecule highly polarized and the intramolecular charge transfer possible. The μ , α and β of 4BAHEHMP are 1.076 4D, 2.671× 10-23 esu and 3.427× 10-30 esu, respectively, obtained by B3LYP/6-311++G (d, p) method. Urea is the prototypical molecule used in the study of the NLO properties of the molecular systems. Therefore urea was used frequently as a threshold value for comparative purposes. In this study, the total dipole moment, polarizability and the calculated β value of the molecule is greater than urea (the μ , α and β of urea are 1.373 2D, 3.831 2× 10-24 esu and 0.372 89× 10-30 esu). Hence the first order hyperpolarizability (β tot) of 4BAHEHMP with B3LYP/6-311++G (d, p) basis set is ten times larger than the urea value. From the computation the high values of 4BAHEHMP are probably attributed to the charge transfer existing between the phenyl rings within the molecular skeleton. We conclude that the compound 4BAHEHMP is an attractive object for future studies of non-linear optical properties.
3 Conclusion
Geometry of 4BAHEHMP molecule was optimized using DFT at B3LYP level of theory employing 6-311++G(d, p) basis set. The calculated values are also compared with experimental data. In general the structural parameters matches well with the experimental one, with few exceptions caused due to constraints imposed by isolated molecule model. A detailed normal coordinate analysis of all the normal modes along with PED very clearly indicates the composition of each normal mode in terms of internal coordinates. HOMO-LUMO, MEP and NBO may serve as a useful quantity to explain hydrogen bonding, reactivity and structure-activity relationship of molecules. Fukui function helps in identifying the electrophilic and nucleophilic nature of a specific site within a molecule. The predicted first order hyperpolarizability shows that the molecule might have a reasonably good nonlinear optical (NLO) behavior. The correlations between the statistical thermodynamics and temperature are also obtained. It was seen that the heat capacities, entropies and enthalpies increase with the increasing temperature owing to the intensities of the molecular vibrations increase with increasing temperature.
The authors have declared that no competing interests exist.
参考文献
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|