
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HNIW和DNT的共晶制备和光谱分析研究
[刘可1, 2  , 张皋
, 张皋1, 2 , 陈智群1, 2 , 舒远杰1, 2 , 栾洁玉1, 2 ]
, 张皋|
|


作者简介: 刘 可, 女, 1992年生, 西安近代化学研究所助理工程师 e-mail: happycoco5133@163.com
六硝基六氮杂异伍兹烷(HNIW)作为一种新型高能炸药, 寻求对其综合性能的改性是现阶段研究的重点。 不同于传统的重结晶、 高聚物包覆和复合等改性方法, 通过晶体工程学方法, 采用和小分子低感度炸药共结晶的方法, 可对HNIW从炸药的内部组成和晶体结构层面进行改性。 设计采用溶剂挥发法以一定的配比制备得到了六硝基六氮杂异伍兹烷(HNIW)和二硝基甲苯(DNT)的共晶, 并针对晶体结构以及物相的改变, 应用X射线粉末衍射(PXRD)、 傅里叶变换红外光谱(FTIR)和拉曼光谱(Raman)相结合的方法, 对共晶样品进行快捷而有效的表征。 结果显示, 与单组分HNIW和DNT相比, 共晶的PXRD图谱中出现了新的强衍射峰, 证实了共晶与单组分晶体结构具有显著差异, 产生了一种新的物相。 红外光谱和拉曼光谱谱图显示, 一系列特征吸收峰产生了峰位偏移以及峰宽和峰高的变化, 证明了由于HNIW分子和DNT分子之间相互作用形成了共晶分子晶体结构, 且共晶内部晶格振动较之单组分HNIW和DNT发生了改变, 不再是单组分分子间的相互作用, 从而佐证了HNIW/DNT共晶是一种不同于原组分分子空间构型的新物相。
As a new high energy explosive, it is particularly crucial for HNIW (2, 4, 6, 8, 10, 12-hexanitrohexaazaiso-wurtzitane) to modify the comprehensive performance especially under the modern battlefield environment. Unlike the ordinary modifying method such as recrystallization and polymer coating, the formation of HNIW cocrystal with low sensitivity micromolecule explosive can achieve the modification from the inner composition and the invariant inherent molecular structure. Consequently, HNIW/DNT(dinitrotoluene) cocrystal was prepared by slow solvent evaporation method at room temperature. Using X-ray diffraction, Fourier transform infrared spectroscopy (FTIR) and Raman spectroscopy analysis, the crystal structure, intermolecular non-bonding interactions and comprehensive properties of the cocrystal were characterized and analyzed. The characterization results showed that a new sharp peak appeared in the co-crystal PXRD diffraction pattern, which indicated that the cocrystal was a totally new non-solvated crystalline phase rather than the crystal transformation product. In fourier transform infrared spectroscopy and Raman spectroscopy, the peak shifts and peak height, peak width of some characteristic absorption peak changed, which confirmed the formation of HNIW/DNT cocrystal. And the intermolecular interactions was mainly the result of intermolecular interactions between HNIW and DNT molecular.
六硝基六氮杂异伍兹烷(HNIW, 又称CL-20)是一种新型立体笼状多硝铵化合物, 1987年由美国海军武器中心(NWC)的Nielsen[1]等首次合成, 是目前能量最高、 综合性能最为良好的单质炸药之一, 其在高性能、 低特征信号推进剂及高能混合炸药中的应用, 已经进行深入而系统的研究[2, 3, 4]。 现有针对HNIW的改性如重结晶、 高聚物包覆和复合等方法, 均未使其内部组成和晶体结构产生改变, 因此迫切需要其他有效改性途径。 其中通过晶体工程学方法, 基于各组分的分子识别、 组装、 分子间设计与其他组分合成共晶炸药更是引起了研究人员的重视[5, 6, 7]。 共晶体系是分子(两种或者两种以上)通过氢键等分子间相互作用, 形成具有特定结构和性能的多组分分子晶体, 实质上为不同分子间相互作用的结果, 其在热力学、 动力学、 分子识别平衡的基础上, 可以得到结构可控、 具有特定物化性质的新晶体[7, 8, 9, 10]。 应用到HNIW的改性上, 可以通过对晶体内分子非键作用(氢键、 π — π 共轭作用、 卤键等)进行分子设计, 将HNIW和其他低感度组分复合形成共晶炸药, 在笼状的HNIW大分子中插入其他小分子, 微观分子层面实现更强的分子间非键作用和更致密的晶体结构, 可以在不破坏原有材料分子化学结构和化学键的前提下, 根本上改变晶体内部组成和晶体结构, 大大拓展现有单质HNIW的应用范围。 沈金朋等[11]采用拉曼光谱和X射线粉末衍射综合表征HMX/TATB共晶, HMX中的硝基与TATB中氨基之间可以形成三种类型的N— O…H氢键, 依靠氢键相互作用结合形成共晶, 与之前分子动力学理论模拟方法设计的HMX/TATB共晶结构相吻合。 郭长艳等[12]同样用溶剂挥发法合成了BTF和TNT等七种炸药的共晶, 利用粉末X射线衍射进行表征证实了其中五种体系共晶的形成。
针对共晶表征的关键是证实形成共晶的两种组分分子间相互作用(氢键、 π — π 共轭作用等)的形成, 最为准确是单晶X射线衍射, 可以得到晶体微观结构三维图和晶胞常数, 但测试样品必须是单晶, 且对尺寸、 结晶度和光泽度都有所要求。 相比之下, 利用傅里叶变换红外光谱(FTIR)和拉曼光谱结合的方法, 通过检测并比照共晶和单组分分子中特征吸收峰峰位、 峰宽等特性的变化, 可有效探测晶格内部分子的振动。 此外结合X射线粉末衍射(PXRD), 通过对照衍射图上的晶体衍射谱线(衍射峰数目、 位置和强度), 也能够判定是否产生了新的物相和晶体结构。 基于以上分析, 针对所制备的六硝基六氮杂异伍兹烷(HNIW)和二硝基甲苯(DNT)的共晶, 采用红外光谱、 拉曼光谱和X射线粉末衍射三种方法进行表征, 对共晶的内部分子间作用力进行研究。
实验所用六硝基六氮杂异伍兹烷和二硝基甲苯均来自兵器工业805厂, 并经过精制, 品级为分析纯, 使用前未进行进一步纯化。 所用溶剂乙酸乙酯, 生产厂家为广州光华化学试剂有限公司, 品级为分析纯。
将六硝基六氮杂异伍兹烷和二硝基甲苯按照1:2的计量比, 各自称量484和216 mg, 溶于5 mL乙酸乙酯中, 超声振荡混合均匀至固体完全溶解后, 将溶液转移至锥形瓶中, 置于阴凉通风处, 常温下溶剂缓慢挥发得到淡黄色透明晶体, 将析出的晶体洗涤干燥, 得到共晶。
X射线粉末衍射分析采用德国Bruker公司制造的SMART型号衍射仪, Cu-Kα 射线, 工作电压为40 kV, 工作电流40 mA, 扫描角度范围为5° ~90° , 步长0.02° /0.1 s。 红外光谱分析采用美国Thermo公司制造的NEXUS型傅里叶变换红外光谱仪, 仪器厂家为Thermo-Fisher, 型号为NEXUS, 样品采用KBr压片法, 仪器分辨率为4 cm-1; 拉曼光谱分析采用inVia公司制造的RENISHAW型激光显微拉曼光谱仪, 使用激光光源波长为785 nm, 功率为250 mW, 分辨率为1 cm-1。
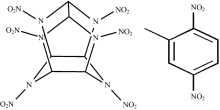
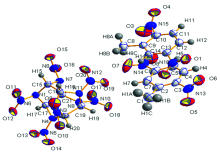
HNIW和DNT的分子结构如图1所示, DNT分子和HNIW分子中都含有硝基基团, HNIW分子中含有笼形结构, DNT分子中含有苯环和甲基等官能团, 分析共晶的分子结构如图2所示, 两种分子间可能存在C— H…O氢键作用(DNT的硝基氧原子和HNIW环上氢原子, DNT甲基氢原子和CL-20硝基氧原子之间), 以及芳香环之间的π — π 共轭作用。 由于共晶分子中分子间弱相互作用的影响, 原有化学键和官能团在光谱中的特征吸收峰以及衍射图谱中的衍射峰将会产生不同程度的偏移和改变。
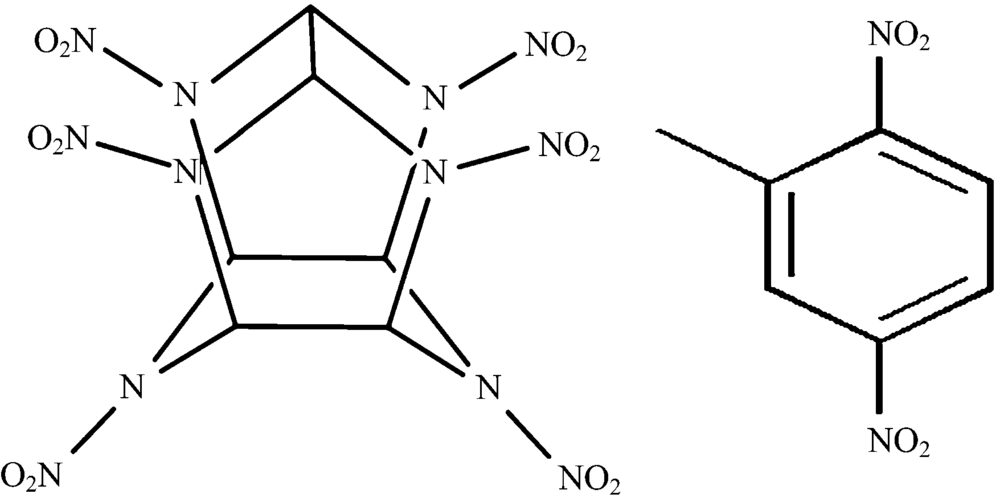 | 图1 HNIW和DNT分子结构Fig.1 Molecular structures of HNIW and DNT |
 | 图2 HNIW/DNT共晶分子结构图Fig.2 Cell unit of HNIW/DNT cocrystal |
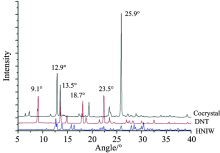
HNIW和DNT共晶以及两种单组分的PXRD图谱如图3所示, 很明显共晶的衍射图谱与单组分HNIW和DNT有着较大的差异, 最为显著的是共晶PXRD图谱中在25.9° 处出现一个极强的衍射峰, 峰强度大且峰形尖锐, 这是在HNIW和DNT衍射图谱中都没有出现的。 另外在12.9° 处产生了一个次强衍射峰, 原有DNT谱线中9.1° 和23.5° 处的衍射峰也在共晶图谱中消失了。 如果试样仅为DNT和HNIW的混合物, 所得到的衍射图谱则会是两种单组分晶体的衍射图谱的加和, 并不会因为物质的混聚产生峰位和峰强的变化。 因此衍射图谱一系列的差异说明共晶的晶体结构较之HNIW和DNT的发生了根本改变, 产生了一种新的物相, 从而证明了共晶的形成。
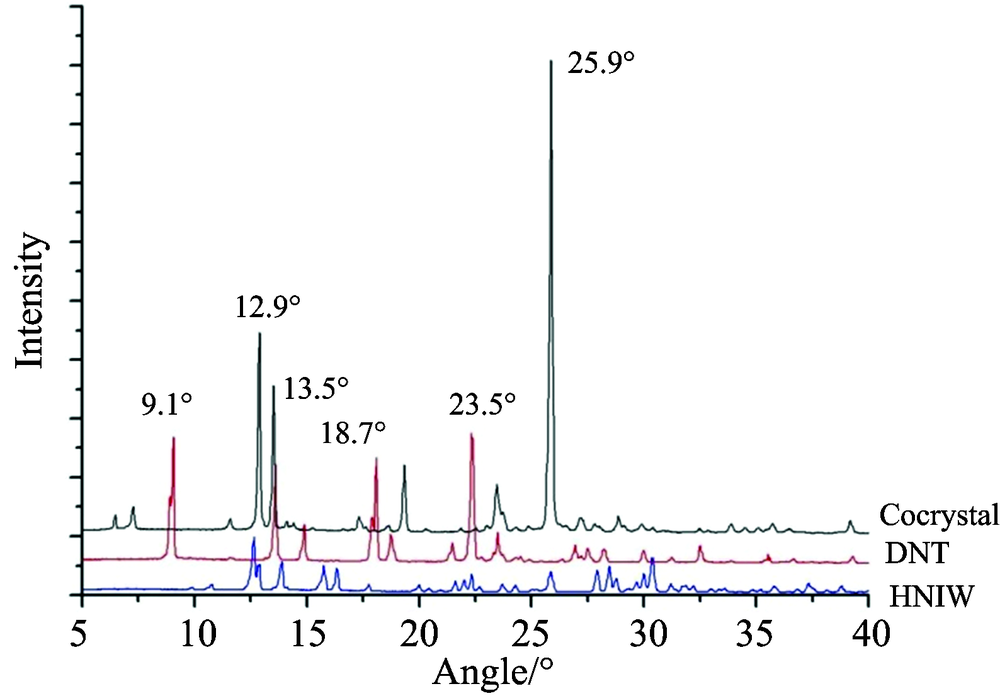 | 图3 HNIW, DNT及共晶的PXRD图谱Fig.3 PXRD patterns of HNIW, DNT and HNIW/DNT cocrystal |
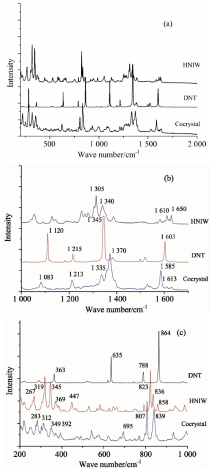
共晶以及HNIW, DNT单组份的红外光谱如图4(a)所示, 为了更清晰地观察共晶与单组份各个吸收峰的差异, 分别将800~1 700 cm-1的指纹吸收振动区域, 以及2 500~4 000 cm-1的C— H振动区域展开放大进行观察, 如图4(b)和图4(c)所示。 由三种红外谱线的对照可以发现, 共晶的各个吸收峰都产生了一些不等的位移现象, 具体如: 共晶的— NO2对称伸缩系列吸收峰位为1 630, 1 602, 1 591和1 572 cm-1, 而DNT和HNIW的— NO2对称伸缩系列吸收峰分别为1 533, 1 612, 1 605, 1 589和1 566 cm-1; 共晶的C— H伸缩振动峰峰位为3 034 cm-1, DNT和HNIW的C— H伸缩振动峰峰位为3 099和3 046 cm-1; 共晶的— NO2不对称伸缩振动系列吸收峰峰位为1 353和1 276 cm-1, DNT和HNIW的NO2不对称伸缩系列吸收峰峰位分别为1 329, 1 284和1 352 cm-1。 分析这些吸收峰峰位的差异, 共晶的红外光谱并非HNIW和DNT的加和, 而是较之两种单组分主要吸收峰分别产生了5~10 cm-1的峰位偏移, 分析是由于共晶中HNIW和DNT结构之间存在弱相互作用(分子间氢键以及芳香环之间的π — π 共轭作用), 但C— H…O氢键作用的极性较之N— H…O, O— H…O等氢键要弱很多, 因此并不会在红外谱图中产生明显的吸收峰, 但这种较弱氢键的作用会对分子的化学环境和电子云分布等产生影响, 使得原有化学键在红外谱图中的吸收峰产生峰位偏移、 峰宽和峰面积改变。
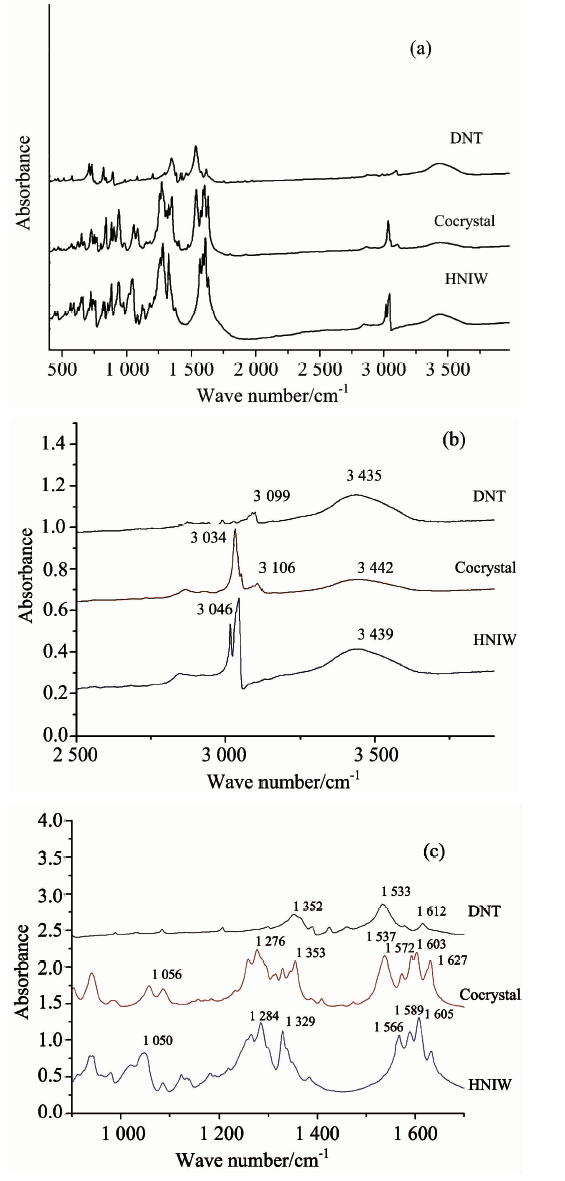 | 图4 HNIW, DNT及共晶红外光谱 (a): 400~4 000 cm-1; (b): 2 500~4 000 cm-1; (c): 800~1 700 cm-1Fig.4 IR spectras of HNIW, DNT and DNT/HNIW cocrystal (a): 400~4 000 cm-1; (b): 2 500~4 000 cm-1; (c): 800~1 700 cm-1 |
共晶以及HNIW, DNT单组分的拉曼光谱如图5(a)所示, 为了更清晰地观察共晶与单组分各个散射峰的差异, 分别将200~1 000 cm-1波长范围, 以及1 000~1 700 cm-1波长范围分别放大如图3(b)和(c)所示。 由拉曼谱图的对照可以发现, 共晶分子的拉曼峰和HNIW和DNT分子的峰有了明显的拉曼位移, 一些特征峰出现了减弱乃至消失的变化。
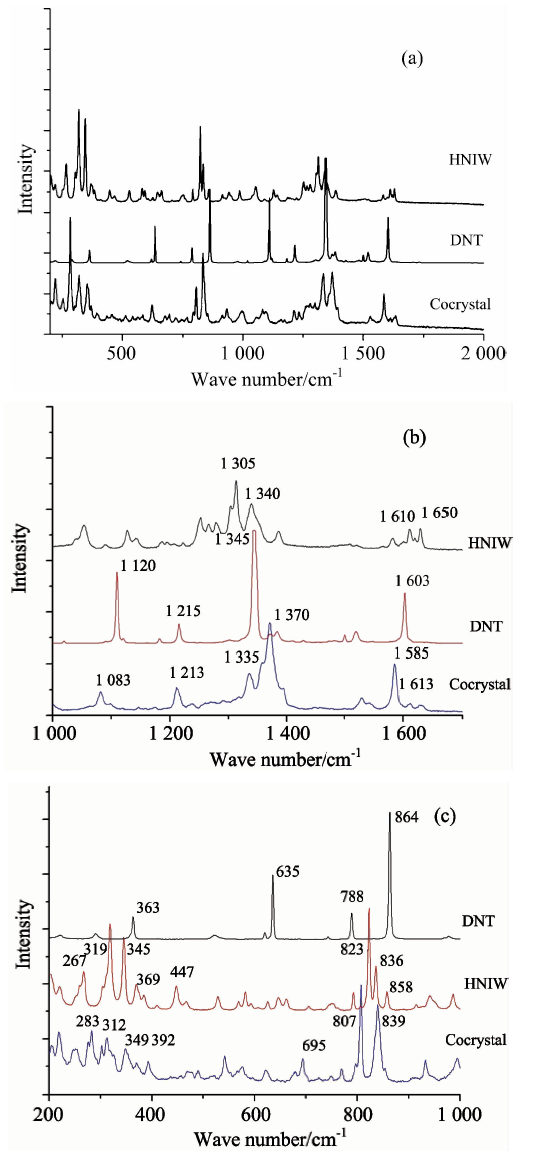 | 图5 HNIW, DNT及共晶拉曼光谱 (a): 400~4 000 cm-1; (b): 2 500~4 000 cm-1; (c): 800~1 700 cm-1Fig.5 Raman spectras of HNIW, DNT and HNIW/DNT cocrystal (a): 400~4 000 cm-1; (b): 2 500~4 000 cm-1; (c): 800~1 700 cm-1 |
具体为: 700~800 cm-1 NO2弯曲振动区域, 共晶的两个较强峰峰位为807和839 cm-1, 并没有出现DNT谱图中864 cm-1的强峰, 较之HNIW的826和836 cm-1也有所偏移; 共晶的C— N键伸缩振动峰峰位为1 585和1 613 cm-1, 而HNIW和DNT的C— N键伸缩振动峰峰位为1 610, 1 650和1 603 cm-1; 共晶的NO2系列伸缩振动峰峰位为1 335和1 370 cm-1, 且其中1 370 cm-1处的峰最宽且最强, 而HNIW和DNT的NO2伸缩振动峰峰位分别为1 305, 1 340和1 345 cm-1; 共晶的C— H键伸缩振动峰峰位为1 083 cm-1, HNIW和DNT的NO2伸缩振动峰峰位分别为1 305, 1 340和1 345 cm-1。
这些主要极性化学键拉曼散射峰峰位及峰形的变化, 与红外光谱中峰位变化的机理基本一致, 都是由于分子间弱相互作用(分子间氢键以及芳香环之间的π — π 共轭作用)所导致。 值得关注的是在200~1 000 cm-1波长范围内, 300 cm-1附近的晶格振动峰, 共晶的峰位为283和312 cm-1, HNIW峰位为267和319 cm-1, DNT则在这一区域没有明显出峰, 进一步证明了共晶内部晶体结构和晶格振动较之单组分HNIW和DNT发生了改变, 不再是单组分分子间的相互作用, 从而佐证了HNIW/DNT共晶是一种不同于原组分分子空间构型的新物相。
采用X射线粉末衍射(PXRD)、 傅里叶变换红外光谱和拉曼光谱相结合的分析方法, 对溶剂挥发法制备所得的六硝基六氮杂异伍兹烷和二硝基甲苯共晶进行表征。 结果显示在X射线粉末衍射图谱中有新的强衍射峰出现, 证实共晶作为一种新的物相的形成, 并与两种单组分晶体结构有所差异。 另外红外光谱和拉曼光谱分析结果表明, HNIW/DNT共晶一系列化学键的特征吸收峰, 包括— NO2伸缩振动峰、 C— N伸缩振动峰等都较之HNIW和DNT分子产生了较大波数的位移, 从而佐证了由于共晶内部组分间作用的形成以及晶体结构的变化。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|