

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
几种香豆素衍生物核磁共振谱的理论研究
[贾飞云1 , 苏奉发1 , 冉鸣2 , 张波1, *  ]
]
]
|
|


在B3LYP / 6-311G(d,p)水平下对六种香豆素衍生物的结构进行了优化, 并通过振动分析验证了其稳定性。 利用GIAO方法在B3LYP/6-311G(d,p)水平下研究了香豆素衍生物的NMR谱。 研究表明, 六个化合物结构共平面性较好, 具有较大共轭体系; 不同的取代基和取代基的不同位置对几种香豆素衍生物的NMR都有不同的影响。 苯环上的氢原子被其他基团取代后, 其α-C原子和邻位碳原子的 δ值变化明显, 而间位碳原子 δ值几乎没有变化。 取代基的电负性对α-C原子 δ值的影响具有一定规律性, 而共轭效应对苯环碳原子的影响则更为复杂。 最后, 为了说明理论计算值与实验值之间的相关性, 给出了六种衍生物化学位移值的理论与实验值相关图, 并进行线性回归。 结果表明, 六种香豆素衍生物的化学位移理论与实验值之间的相关性非常好。
The structure of the six kinds of coumarin derivatives has been optimized at the level of B3LYP/6-311G (d, p), under which the stability has been verified by means of Vibration analysis. Moreover, NMR Spectra of the coumarin derivatives compounds have been studied at the level of B3LYP/6-311G (d, p) by GIAO method. The results showed that the structure of the six compounds, a larger conjugated system, had good planarity. Different Substituents and different positions of substituents all have different influences on NMR of the several coumarin derivatives. In general, after the hydrogen atom on the benzene ring is substituted by other groups, the δ value of the α-C atom next to the substituent changes obviously, the δ value of the Ortho carbon atoms have great change too, but the δ value of the meta carbon atom have almost no change. The effect of electronegativity of substituents on α-C atoms presents obvious regularity, while the influence of conjugate effect on carbon atoms of benzene ring is more complex. Finally, in order to show the correlation between theoretical calculation and experimental values, we showed the correlation diagram of the chemical shift values of the six kinds of derivatives, and carried on the linear regression. Results showed that the correlation between the chemical shift theory and the experimental value of the six kinds of the Coumarin derivatives was very good.
Coumarin and its derivatives are a class of natural products with benzo-α -pyrone nucleus. In structure, it can be seen as the lactone compounds formed by dehydration of cis o-hydroxy cinnamic acid. It is widespread in the higher plants of Rutaceae, Apiaceae, Asteraceae, Leguminosae, Solanaceae, Thymelaeaceae and animal, microbial metabolites. Its derivatives possess obvious biological activities, such as anti-tumor, anti-HIV, anti-cell proliferation, anti-virus, anti-fungal, anti-bacterial, anti-vascular sclerosis, anti-oxidation, enhancing human immunity and so on[1, 2, 3, 4, 5, 6]. In recent years, the structure modification of coumarin derivatives as the lead compound has become a hot research topic. Nuclear magnetic resonance (NMR) spectroscopy is very important tool in areas of chemistry, biology and medicine. Especially in organic chemistry, for some compounds, it is difficult to prepare their single crystals. Their structure and geometries are often determined by their NMR spectra. From the 1970’ s, quantum chemical calculation for NMR has been becoming one of fascinating areas of researches. Especially recently, with the development of the calculate method and computer technology, quantum chemical calculation for NMR has made great progress[7, 8, 9, 10]. With quantum chemical calculation for NMR, this paper discusses the influence of different Para-substituent on 13C NMR of the coumarin derivatives.
The geometric structure of six kinds of the coumarin derivatives has been optimized at the level of B3LYP/6-311G (d, p), under which the stability of the geometric structure has been verified by means of vibration analysis. Moreover, NMR Spectra of the coumarin derivatives compounds has been studied at the level of B3LYP/6-311G(d, p) by gauge including atomic orbital (GIAO) method. All computational work is completed by Gaussian 09 program.
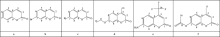
Fig.1 lists the six compounds structure and different substituent. In order to facilitate the discussion, we only numbered the carbon atoms in all compounds, and kept the same number of carbon atoms in the same position.
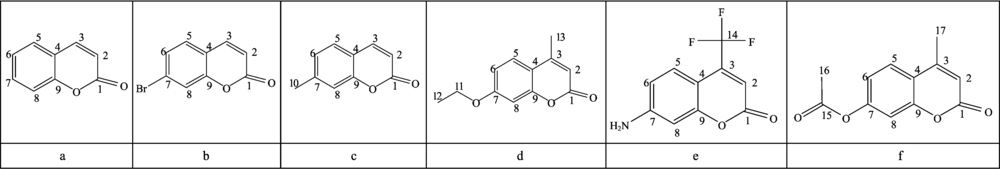 | Fig.1 Structural formulas of the six compounds |
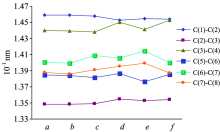
Table 1 lists the geometry structure parameters of the six compounds. In order to be more intuitive to see the change in bond lengths and bond angles, we have drawn the Fig.2 and Fig.3 that represented a change trend about part of bond angles and bond lengths. From the torsion angle data in Table 1, it is shown that the main structure of the six compounds is nearly in the same plane, forming a larger conjugated system. Among them, because of the influences of acetoxy and amino, the coplanarity of benzene ring worsened slightly. Such as torsion angle in compound-f, ∠C(6)C(7)C(8)O=176.304° ; in compound-e, ∠C(6)C(7)C(8)N=177.584° .
| Table 1 Selected structure parameters of the six compounds [bond length: 10-1 nm; bond angle and torsion angle :/(° )] |
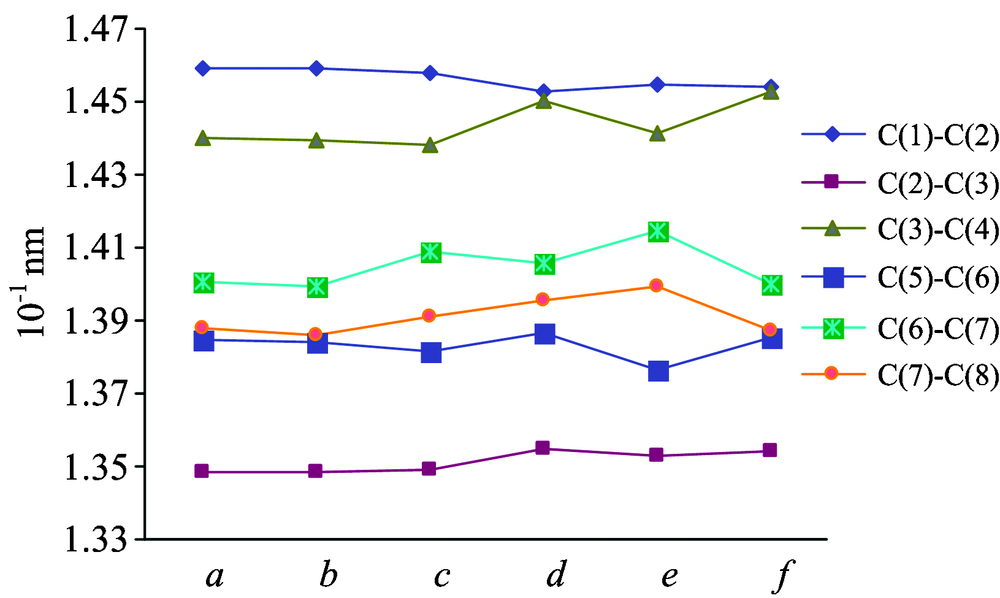 | Fig.2 Influence of different substituent on bond lengths |
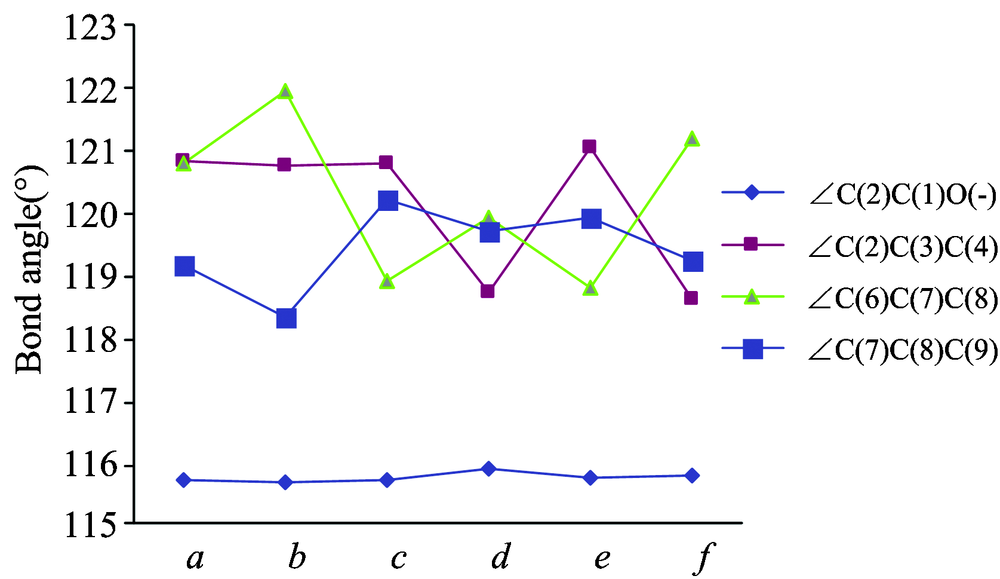 | Fig.3 Influence of different substituent on bond angles |
In Fig.2, from bond lengths of the benzene ring to see, the bond length variation of C(5)-C(6) and C(2)-C(3) is not obvious, and the bond length of C(6)-C(7) and C(7)-C(8) has an obvious change. The reason is probably that the substituents on C(7) have a greater impact on Ortho-position C(6)-C(7) and C(7)-C(8) than Meta-position C(5)-C(6). In general, the conjugation effect, the steric hindrance effect and the electric negative of the neighboring groups are the important factors which affect the bond length of the covalent compounds. From the Fig.2, we can find that the bond length of C(6)-C(7) or C(7)-C(8) in compound-c and compound-e is longer than other compounds. It is reason that methyl group (the substituent in compound-c) is electron-donating groups, and the electron density is biased to the benzene ring; amino groups (the substituent in compound-e) can form π — π conjugated system with the benzene ring, and the electron density is also biased to the benzene ring. They can all activate the benzene ring, resulting in longer bond length. In compound-b, although the substituent (-Br) is electron withdrawing groups, it can also form π — π conjugated system with the benzene ring, so its bond length of C(6)-C(7) and C(7)-C(8) is almost constant. Furthermore, as shown in Table 1, all the compounds have the narrower bond length range and more average bond lengths, wherein the C— C bond length is slightly shorter than it in Ethane and the C=C bond length is slightly longer than it in Ethylene. So this is further proof of the existence of conjugate system in these compounds.
Fig.3 lists the selected bond angle information of these compounds. It is shown that the variation of bond angle ∠C(6)C(7)C(8) is greater than others, and it in compound-b and -f becomes larger, while it in compound-c and -e becomes smaller, but for bond angles on the ortho ∠C(7)C(8)C(9), the situation is reversed. We think the reason is that different substituents in compounds have different repulsive force to the ortho carbon atom. From Fig.3, it is also shown that the bond angles ∠C(2)C(1)O(-) did not change significantly.
The vibration frequency of the six compounds is calculated at the level of b3lyp/6-311G (d, p). As shown in Table 2, the minimum vibration frequency of the six compounds is positive, with no imaginary frequency, so the geometric
structure optimization is reasonable[11].
| Table 2 The minimum vibrational frequency and intensity for the six compounds |
In Table 3, the data is calculated at the level of b3lyp/6-311G (d, p) by GIAO method. The δ value is the relative chemical shift, which is the difference between the absolute shift of TMS and the calculated shift. The absolute shift of TMS is reported by the paper[12].
| Table 3 Selected calculated and experimental chemical shifts of the six compounds (δ /ppm) |
As shown in Fig.1, the compound-a is a coumarin molecule that has no substituents on the ring, while the compound-b~f are all coumarin derivative and have different substituents on C(3) or C(7).In order to be more intuitive to show the different effects of different substituents on C(3) and C(7) on the chemical shift of each compound, we draw the graph of Fig.4. From Fig.4, it is shown that different Substituents and different positions of substituents all have different influence on NMR of the several coumarin derivatives. Compared with compound-a, the largest change of chemical shift in compound-b~f was C(7), followed by C(3), C(6) and C(8), and the smallest change in chemical shift is C(1), C(5) and C(9). In general, after the hydrogen atom on the benzene ring is substituted by other groups, the δ value of the α -C(C(7)) atom next to the substituent changes obviously; the δ value of the ortho carbon atoms [C(6) and C(8)] have a great change too; the δ value of the meta carbon [C(5) and C(9)] atom have almost no change. In compound-d and f, the oxygen atom is directly connected with the benzene ring. Due to oxygen strong electron withdrawing, the α -C electron cloud density is reduced to the shielding effect, resonance bias low field, the chemical shift value increases. In compound-b, the substituent of -Br on C(7) is electron withdrawing group, with strong electron withdrawing, so the electronic cloud density of carbon atom is [C(7)] directly connected with the substituent reduced. The shielding effect is weaken, the chemical shift is biased to low field and theδ value increased. However, due to the heavy atom effect, with respect to the substitution of -Cl and -F, the substitution of -Br will produce a high field shift for the resonance of α -C. This is why the chemical shift of C(7) is bigger but not too much. In compound-e, the substitution of amino group is the reason for the chemical shift of C(7). Amino is ortho para directing group, and mesomeric effect made resonance of ortho carbon atom on benzene shift to high field. Therefore, the chemical shift values of C(6) and C(8) are significantly smaller in the compound-e.Substituents on C(3) are alkyl groups. Substituted alkyl groups make chemical shifts of the substituted carbon atom corresponding increase and due to the influence of the space effect, the larger the alkyl group, the more branches, the greater the chemical shift value of the substituted carbon atom. The electron withdrawing of the carbonyl oxygen atom makes the carbonyl carbon atom in the electron deficient state, so the resonance is in the low field. But when the carbonyl group is connected with the impurity atom or the unsaturated group, the electron shortage of the carbonyl carbon atom can be relieved by the conjugated effect or the inducing effect, resonance is biased to high field and the δ value decreased.
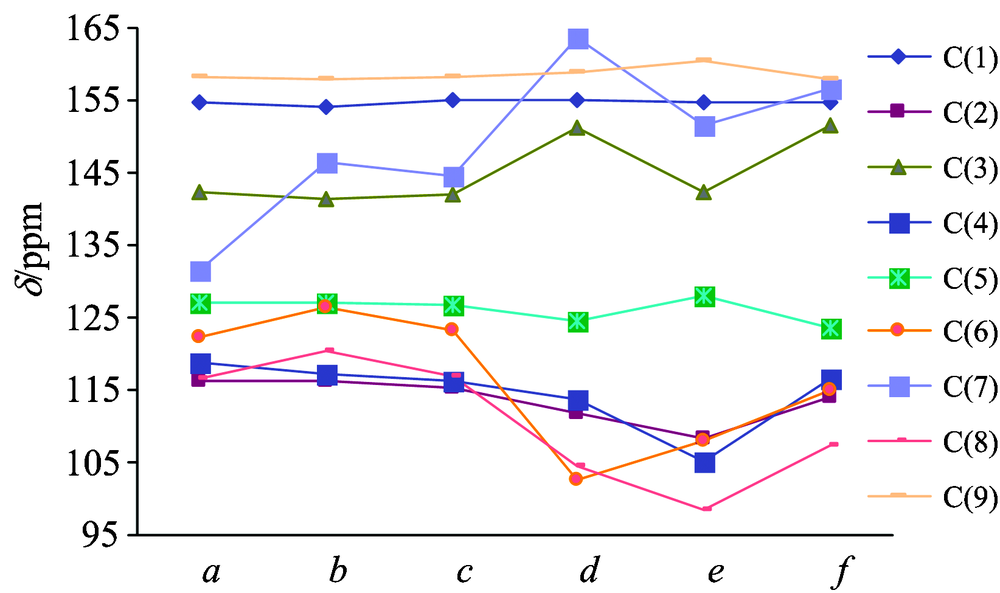 | Fig.4 Selected calculated chemical shifts of the six compounds (δ /ppm) |
As shown in table 3, the δ (about 160 ppm) value of C(1) is smaller than it (about 201 ppm) in acetaldehyde.
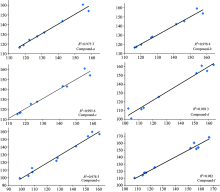
In order to show the correlation between theoretical calculation and experimental values, we take the theoretical δ value for the X axis, the experimental δ value for the Y axis, showing the correlation diagram of the chemical shift values of these six kinds of derivatives, and we carry on the linear regression. The correlation coefficient value is on the chart. Figure 5 shows that the correlation between the chemical shift theory and the experimental value of the six kinds of the Coumadin derivatives is very good, and the correlation values (R2) are 0.975 3, 0.978 4, 0.995 6, 0.969 3, 0.978 5 and 0.982 0 respectively.
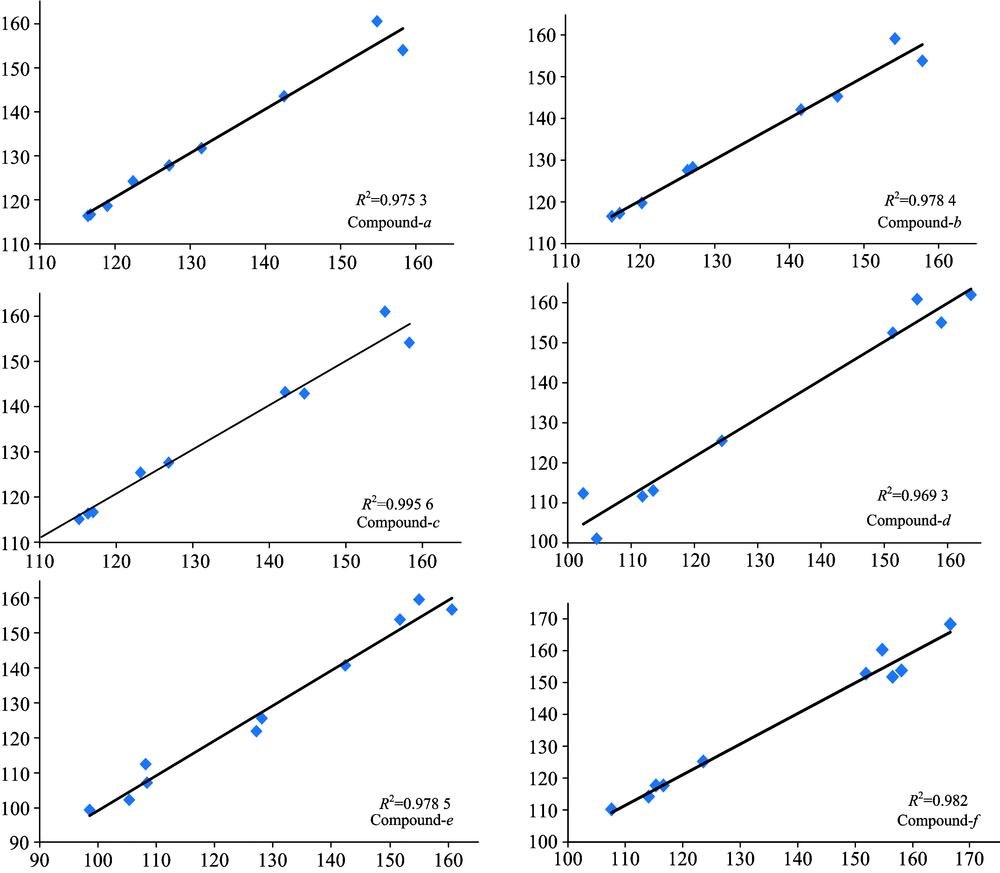 | Fig.5 Correlation of calculated and experimental chemical shifts (13C NMR) of the six compounds |
The main structure of the six compounds was nearly in the same plane, forming a larger conjugated system. Because of the influence of acetoxy and amino, the coplanarity of benzene ring worsened slightly. The conjugation effect, the steric hindrance effect and the electric negative of the neighboring groups are the important factors which affect the bond length of the covalent compounds. Different Substituents and different positions of substituents all have different influence on NMR of the several coumarin derivatives. Substituted alkyl groups make chemical shifts of the substituted carbon atom corresponding increase and due to the influence of the space effect, the larger the alkyl group, the more branches, the greater the chemical shift value of the substituted carbon atom. The effect of electronegativity of substituents on α -C atoms presents obvious regularity, while the influence of conjugate effect on carbon atoms of benzene ring is more complex. Finally, in order to show the correlation between theoretical calculation and experimental values, we show the correlation diagram of the chemical shift values of these six kinds of derivatives, and carry on the linear regression. Results show that the correlation between the chemical shift theory and the experimental value of the six kinds of the Coumadin derivatives is very good.
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|